CSPG4 as Target for CAR-T-Cell Therapy of Various Tumor Entities–Merits and Challenges



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
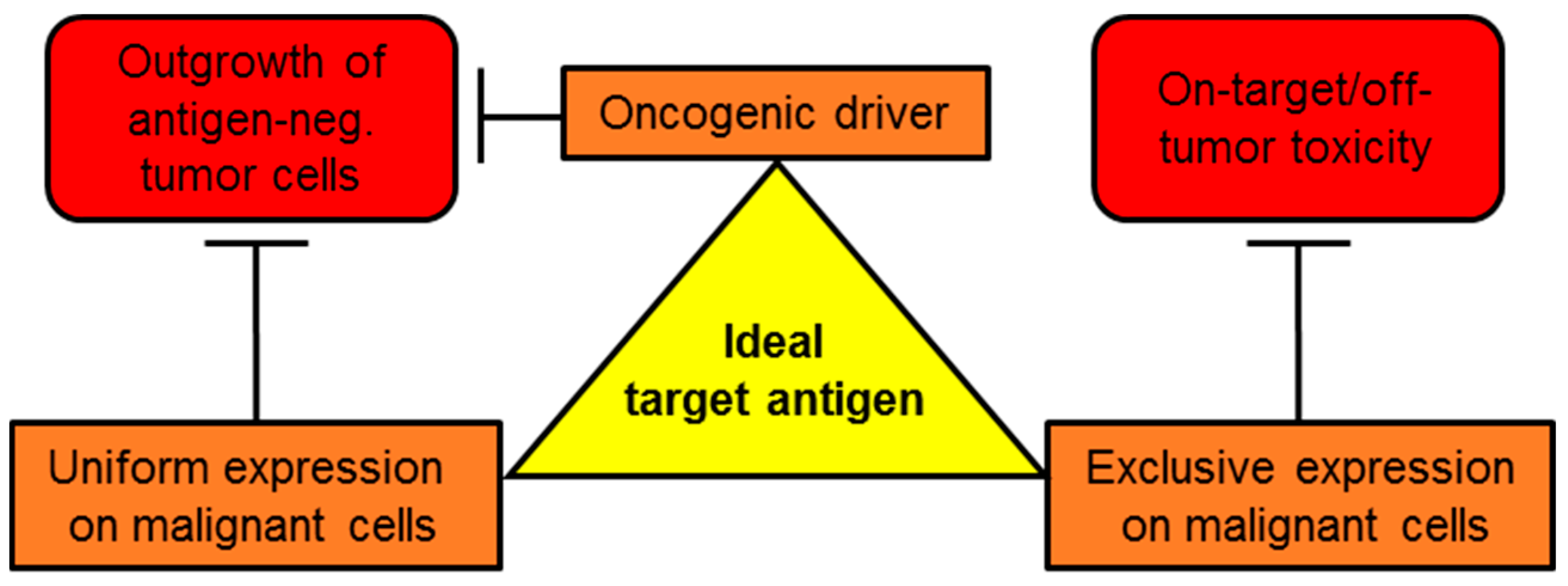
:1. Introduction
2. CSPG4-CAR-T-Cell Therapy of Different Tumors
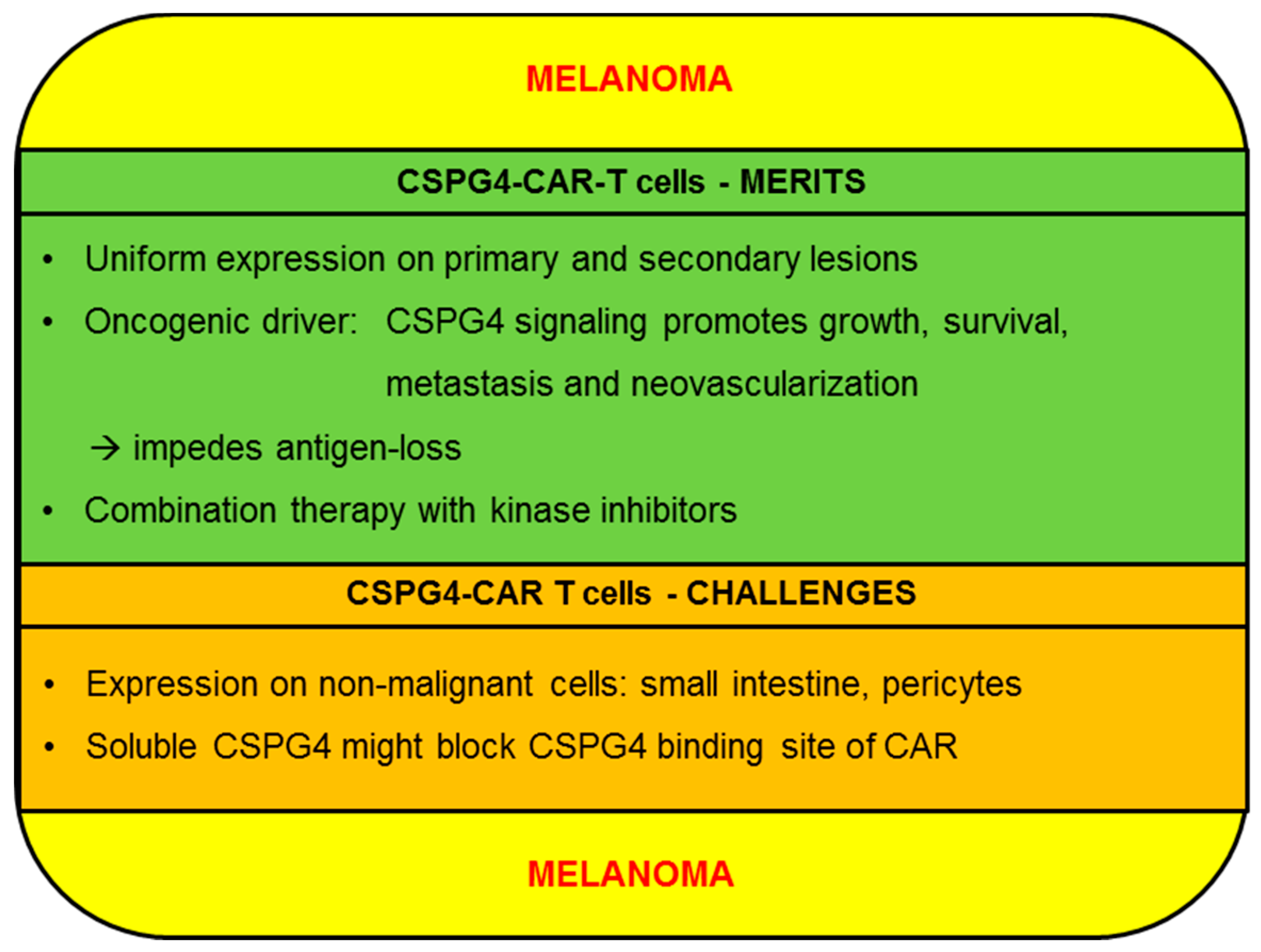
2.1. Melanoma
2.1.1. CSPG4-CAR-T cells: Merits (Melanoma)
2.1.2. CSPG4-CAR-T Cells: Challenges (Melanoma)
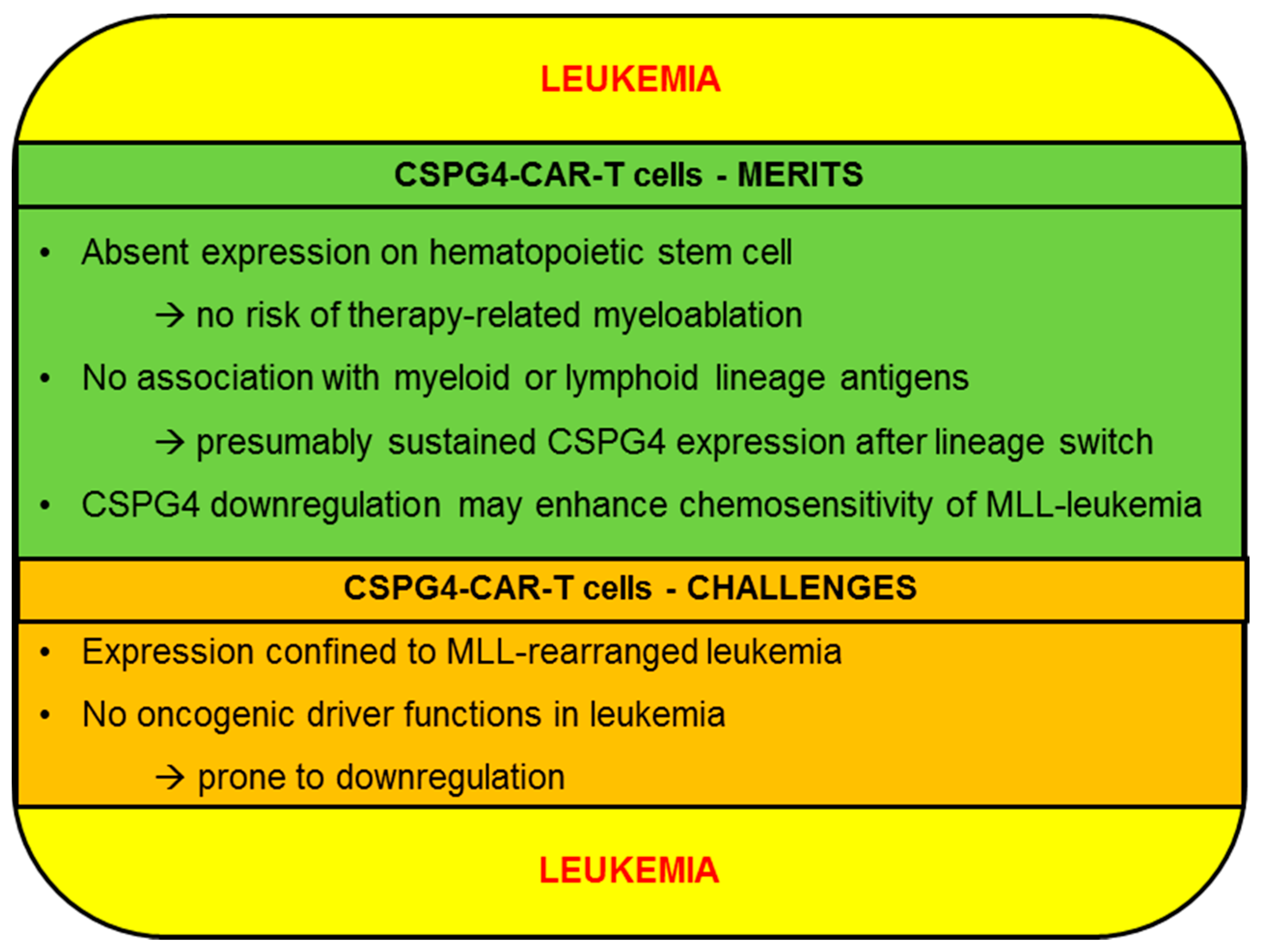
2.2. Leukemia
2.2.1. CSPG4-CAR-T Cells: Merits (Leukemia)
2.2.2. CSPG4-CAR-T Cells: Challenges (Leukemia)
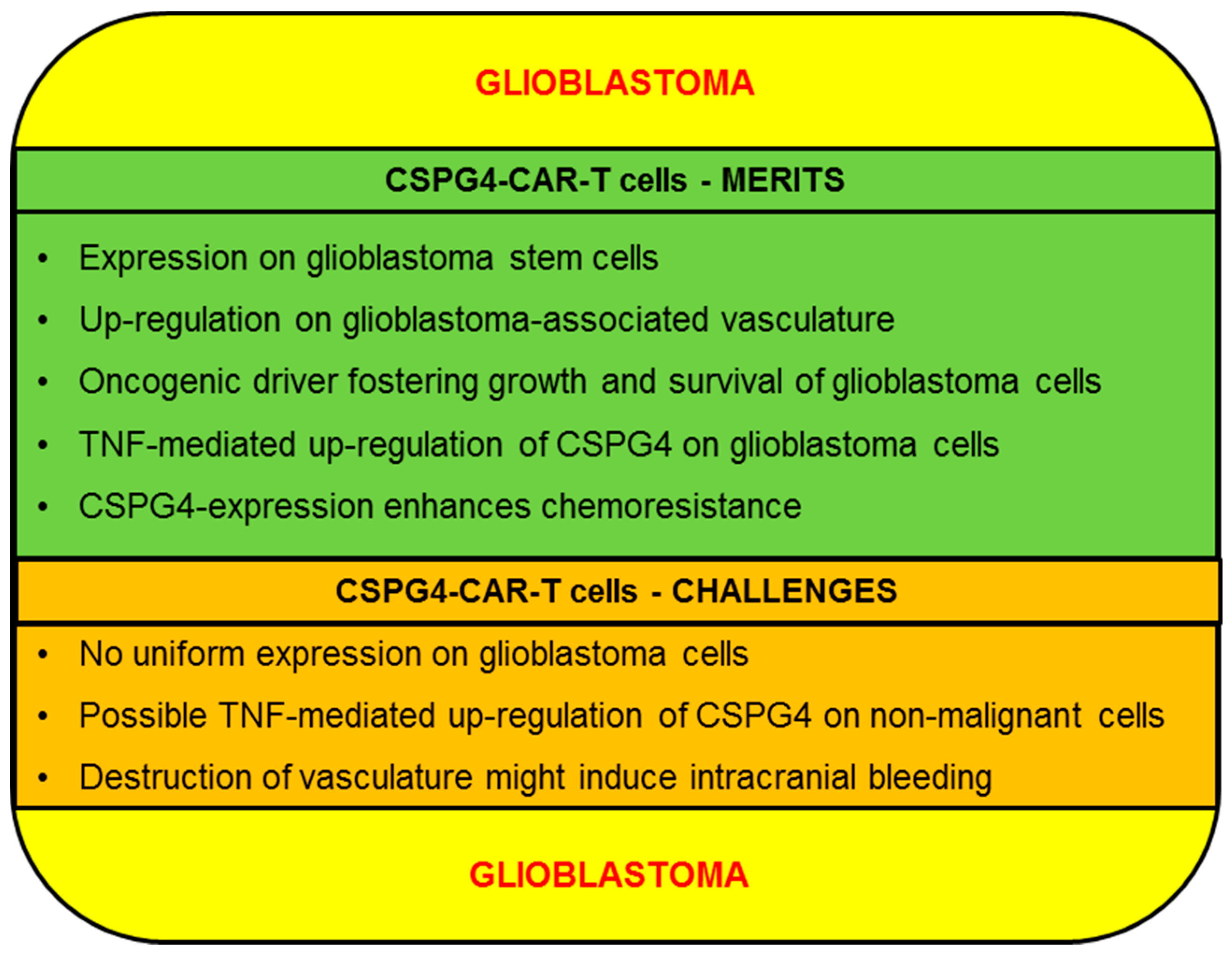
2.3. Glioblastoma
2.3.1. CSPG4-CAR-T Cells: Merits (Glioblastoma)
2.3.2. CSPG4-CAR-T Cells: Challenges (Glioblastoma)
2.4. Triple-Negative Breast Cancer (TNBC)
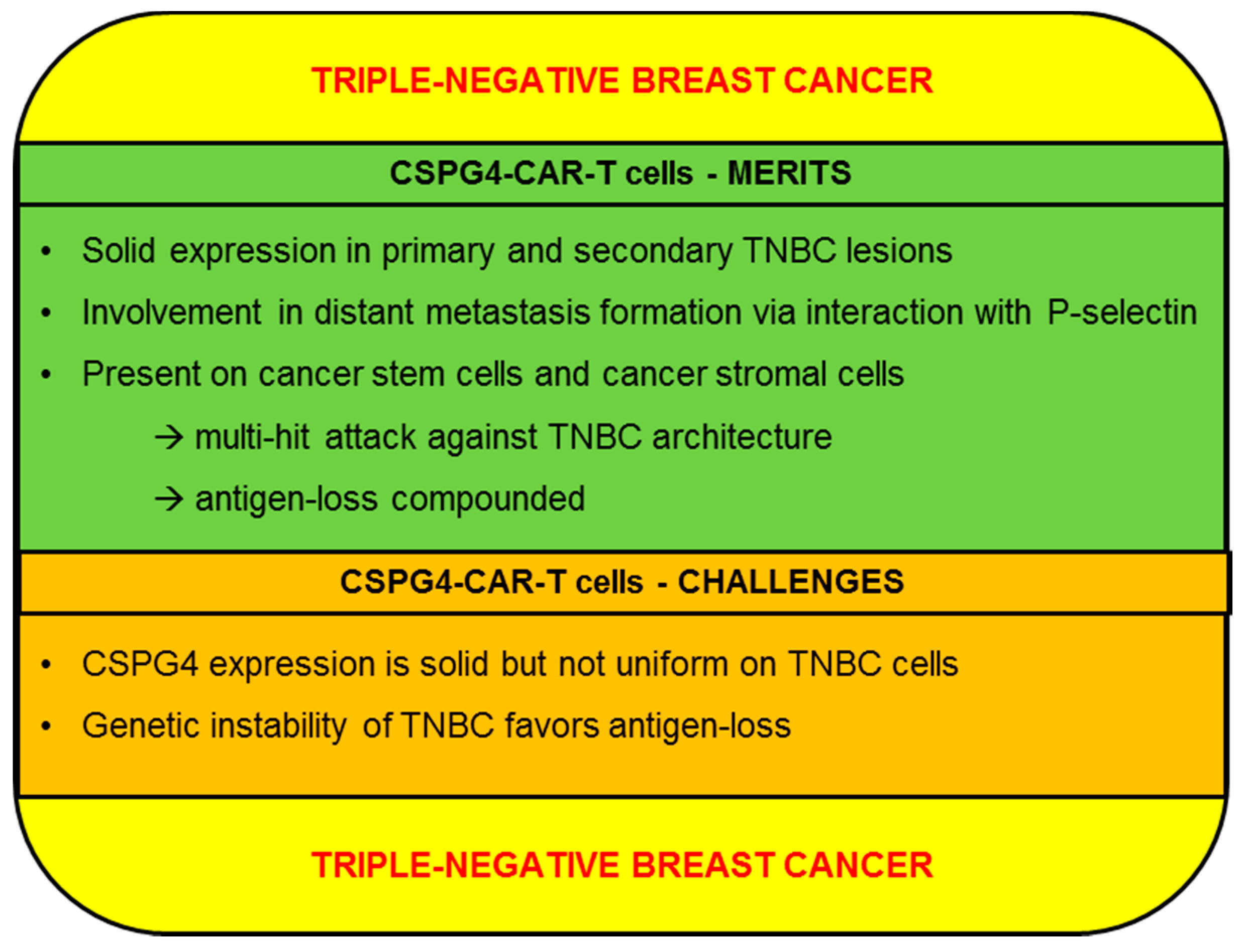
2.4.1. CSPG4-CAR-T Cells: Merits (TNBC)
2.4.2. CSPG4-CAR-T Cells: Challenges (TNBC)
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAR | Chimeric antigen receptor |
| ATT | Adoptive T-cell therapy |
| ALL | Acute lymphoblastic leukemia |
| DLBCL | Diffuse large B-cell lymphoma |
| CSPG4 | Chondroitin sulfate proteoglycan 4 |
| scFv | Single-chain Fragment variable |
| FDA | Food and drug administration |
| EMA | European medicines agency |
| TME | Tumor microenvironment |
| MCSP | Melanoma-associated chondroitin sulfate proteoglycan |
| HMW-MAA | High molecular weight melanoma-associated antigen |
| NG2 | Neural/glial antigen 2 |
| GAG | Glycosaminoglycan |
| CS | Chondroitin sulfate |
| RTK | Receptor tyrosine kinases |
| PKCα | Protein kinase C alpha |
| MAPK | Mitogen-activated-protein-kinase |
| FAK | Focal adhesion kinase |
| TIL | Tumor-infiltrating lymphocyte |
| r/r | refractory and relapsing |
| FLT3 | FMS-like tyrosine kinase 3 |
| FLT3-ITD | FLT3 with internal tandem duplication |
| HSC | Hematopoietic stem cell |
| MLL | Mixed lineage leukemia |
| GBM | Glioblastoma |
| BiTE | Bi-specific T-cell engager |
| GSC | Glioblastoma stem cell |
| EGR1 | Early growth response protein 1 |
| TNBC | Triple-negative breast cancer |
| ER | estrogen receptors |
| EGFR | epidermal growth factor receptor |
| VEGFRvIII | Vascular endothelial growth factor receptor variant III |
| PARP | Poly(ADP-ribose) polymerase |
References
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Go, W.Y. CAR T-Cell Therapy in Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1065. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Subklewe, M.; von Bergwelt-Baildon, M.; Humpe, A. Chimeric Antigen Receptor T Cells: A Race to Revolutionize Cancer Therapy. Transfus. Med. Hemother. 2019, 46, 15–24. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Brentjens, R.J. Tumors evading CARs-the chase is on. Nat. Med. 2018, 24, 1492–1493. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D., Jr.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef]
- Lesch, S.; Benmebarek, M.R.; Cadilha, B.L.; Stoiber, S.; Subklewe, M.; Endres, S.; Kobold, S. Determinants of response and resistance to CAR T cell therapy. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Hartley, J.; Abken, H. Chimeric antigen receptors designed to overcome transforming growth factor-beta-mediated repression in the adoptive T-cell therapy of solid tumors. Clin. Transl. Immunol. 2019, 8, e1064. [Google Scholar] [CrossRef]
- Zhang, E.; Gu, J.; Xu, H. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol. Cancer 2018, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- DeRenzo, C.; Gottschalk, S. Genetic Modification Strategies to Enhance CAR T Cell Persistence for Patients With Solid Tumors. Front. Immunol. 2019, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.S.; Imai, K.; Natali, P.G.; Ferrone, S. Distribution and molecular characterization of a cell-surface and a cytoplasmic antigen detectable in human melanoma cells with monoclonal antibodies. Int. J. Cancer 1981, 28, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.S.; Ruberto, G.; Ferrone, S. Immunochemical characterization of a human high molecular weight--melanoma associated antigen identified with monoclonal antibodies. Cancer Immunol. Immunother. 1983, 14, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Nicolosi, P.A.; Dallatomasina, A.; Perris, R. Theranostic impact of NG2/CSPG4 proteoglycan in cancer. Theranostics 2015, 5, 530–544. [Google Scholar] [CrossRef]
- Campoli, M.; Ferrone, S.; Wang, X. Functional and clinical relevance of chondroitin sulfate proteoglycan 4. Adv. Cancer Res. 2010, 109, 73–121. [Google Scholar]
- Price, M.A.; Colvin Wanshura, L.E.; Yang, J.; Carlson, J.; Xiang, B.; Li, G.; Ferrone, S.; Dudek, A.Z.; Turley, E.A.; McCarthy, J.B. CSPG4, a potential therapeutic target, facilitates malignant progression of melanoma. Pigment Cell Melanoma Res. 2011, 24, 1148–1157. [Google Scholar] [CrossRef]
- Nishiyama, A.; Dahlin, K.J.; Prince, J.T.; Johnstone, S.R.; Stallcup, W.B. The primary structure of NG2, a novel membrane-spanning proteoglycan. J. Cell Biol. 1991, 114, 359–371. [Google Scholar] [CrossRef]
- Fukushi, J.; Makagiansar, I.T.; Stallcup, W.B. NG2 proteoglycan promotes endothelial cell motility and angiogenesis via engagement of galectin-3 and alpha3beta1 integrin. Mol. Biol. Cell 2004, 15, 3580–3590. [Google Scholar] [CrossRef]
- Cooney, C.A.; Jousheghany, F.; Yao-Borengasser, A.; Phanavanh, B.; Gomes, T.; Kieber-Emmons, A.M.; Siegel, E.R.; Suva, L.J.; Ferrone, S.; Kieber-Emmons, T.; et al. Chondroitin sulfates play a major role in breast cancer metastasis: A role for CSPG4 and CHST11 gene expression in forming surface P-selectin ligands in aggressive breast cancer cells. Breast Cancer Res. 2011, 13, R58. [Google Scholar] [CrossRef] [PubMed]
- Makagiansar, I.T.; Williams, S.; Mustelin, T.; Stallcup, W.B. Differential phosphorylation of NG2 proteoglycan by ERK and PKCalpha helps balance cell proliferation and migration. J. Cell Biol. 2007, 178, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Barritt, D.S.; Pearn, M.T.; Zisch, A.H.; Lee, S.S.; Javier, R.T.; Pasquale, E.B.; Stallcup, W.B. The multi-PDZ domain protein MUPP1 is a cytoplasmic ligand for the membrane-spanning proteoglycan NG2. J. Cell Biochem. 2000, 79, 213–224. [Google Scholar] [CrossRef]
- Chatterjee, N.; Stegmuller, J.; Schatzle, P.; Karram, K.; Koroll, M.; Werner, H.B.; Nave, K.A.; Trotter, J. Interaction of syntenin-1 and the NG2 proteoglycan in migratory oligodendrocyte precursor cells. J. Biol. Chem. 2008, 283, 8310–8317. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Lin, X.H.; Giese, N.; Heldin, C.H.; Stallcup, W.B. Interaction between NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells is required for optimal response to PDGF. J. Neurosci. Res. 1996, 43, 315–330. [Google Scholar] [CrossRef]
- Yang, J.; Price, M.A.; Neudauer, C.L.; Wilson, C.; Ferrone, S.; Xia, H.; Iida, J.; Simpson, M.A.; McCarthy, J.B. Melanoma chondroitin sulfate proteoglycan enhances FAK and ERK activation by distinct mechanisms. J. Cell Biol. 2004, 165, 881–891. [Google Scholar] [CrossRef]
- Chekenya, M.; Krakstad, C.; Svendsen, A.; Netland, I.A.; Staalesen, V.; Tysnes, B.B.; Selheim, F.; Wang, J.; Sakariassen, P.O.; Sandal, T.; et al. The progenitor cell marker NG2/MPG promotes chemoresistance by activation of integrin-dependent PI3K/Akt signaling. Oncogene 2008, 27, 5182–5194. [Google Scholar] [CrossRef]
- Ilieva, K.M.; Cheung, A.; Mele, S.; Chiaruttini, G.; Crescioli, S.; Griffin, M.; Nakamura, M.; Spicer, J.F.; Tsoka, S.; Lacy, K.E.; et al. Chondroitin Sulfate Proteoglycan 4 and Its Potential As an Antibody Immunotherapy Target across Different Tumor Types. Front. Immunol. 2017, 8, 1911. [Google Scholar] [CrossRef]
- Schiffer, D.; Mellai, M.; Boldorini, R.; Bisogno, I.; Grifoni, S.; Corona, C.; Bertero, L.; Cassoni, P.; Casalone, C.; Annovazzi, L. The Significance of Chondroitin Sulfate Proteoglycan 4 (CSPG4) in Human Gliomas. Int. J. Mol. Sci. 2018, 19, 2724. [Google Scholar] [CrossRef]
- Van Sinderen, M.; Cuman, C.; Winship, A.; Menkhorst, E.; Dimitriadis, E. The chrondroitin sulfate proteoglycan (CSPG4) regulates human trophoblast function. Placenta 2013, 34, 907–912. [Google Scholar] [CrossRef]
- Sakry, D.; Neitz, A.; Singh, J.; Frischknecht, R.; Marongiu, D.; Biname, F.; Perera, S.S.; Endres, K.; Lutz, B.; Radyushkin, K.; et al. Oligodendrocyte precursor cells modulate the neuronal network by activity-dependent ectodomain cleavage of glial NG2. PLoS Biol. 2014, 12, e1001993. [Google Scholar] [CrossRef] [PubMed]
- Legg, J.; Jensen, U.B.; Broad, S.; Leigh, I.; Watt, F.M. Role of melanoma chondroitin sulphate proteoglycan in patterning stem cells in human interfollicular epidermis. Development 2003, 130, 6049–6063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Svendsen, A.; Kmiecik, J.; Immervoll, H.; Skaftnesmo, K.O.; Planaguma, J.; Reed, R.K.; Bjerkvig, R.; Miletic, H.; Enger, P.O.; et al. Targeting the NG2/CSPG4 proteoglycan retards tumour growth and angiogenesis in preclinical models of GBM and melanoma. PLoS ONE 2011, 6, e23062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Katayama, A.; Wang, Y.; Yu, L.; Favoino, E.; Sakakura, K.; Favole, A.; Tsuchikawa, T.; Silver, S.; Watkins, S.C.; et al. Functional characterization of an scFv-Fc antibody that immunotherapeutically targets the common cancer cell surface proteoglycan CSPG4. Cancer Res. 2011, 71, 7410–7422. [Google Scholar] [CrossRef] [Green Version]
- Wick, M.R.; Swanson, P.E.; Rocamora, A. Recognition of malignant melanoma by monoclonal antibody HMB-45. An immunohistochemical study of 200 paraffin-embedded cutaneous tumors. J. Cutan. Pathol. 1988, 15, 201–207. [Google Scholar] [CrossRef]
- Ramos, C.A.; Savoldo, B.; Torrano, V.; Ballard, B.; Zhang, H.; Dakhova, O.; Liu, E.; Carrum, G.; Kamble, R.T.; Gee, A.P.; et al. Clinical responses with T lymphocytes targeting malignancy-associated kappa light chains. J. Clin. Investig. 2016, 126, 2588–2596. [Google Scholar] [CrossRef] [Green Version]
- Hilden, J.M.; Dinndorf, P.A.; Meerbaum, S.O.; Sather, H.; Villaluna, D.; Heerema, N.A.; McGlennen, R.; Smith, F.O.; Woods, W.G.; Salzer, W.L.; et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: Report on CCG 1953 from the Children’s Oncology Group. Blood 2006, 108, 441–451. [Google Scholar] [CrossRef]
- Yang, L.; Chen, C.J.; Guo, X.L.; Wu, X.C.; Lv, B.J.; Wang, H.L.; Guo, Z.; Zhao, X.Y. Bevacizumab and risk of intracranial hemorrhage in patients with brain metastases: A meta-analysis. J. Neurooncol. 2018, 137, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Warta, R.; Herold-Mende, C.; Chaisaingmongkol, J.; Popanda, O.; Mock, A.; Mogler, C.; Osswald, F.; Herpel, E.; Kustner, S.; Eckstein, V.; et al. Reduced promoter methylation and increased expression of CSPG4 negatively influences survival of HNSCC patients. Int. J. Cancer 2014, 135, 2727–2734. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Tang, Y.J.; Wei, Q.; Hirata, M.; Weng, A.; Han, I.; Okawa, A.; Takeda, S.; Whetstone, H.; Nadesan, P.; et al. Mesenchymal Tumors Can Derive from Ng2/Cspg4-Expressing Pericytes with beta-Catenin Modulating the Neoplastic Phenotype. Cell Rep. 2016, 16, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, E.M.V.; Lindberg, M.F.; Jespersen, H.; Alsen, S.; Bagge, R.O.; Donia, M.; Svane, I.M.; Nilsson, O.; Ny, L.; Nilsson, L.M.; et al. HER2 CAR-T Cells Eradicate Uveal Melanoma and T-cell Therapy-Resistant Human Melanoma in IL2 Transgenic NOD/SCID IL2 Receptor Knockout Mice. Cancer Res. 2019, 79, 899–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallstabe, L.; Mades, A.; Frenz, S.; Einsele, H.; Rader, C.; Hudecek, M. CAR T cells targeting alphavbeta3 integrin are effective against advanced cancer in preclinical models. Adv. Cell Gene Ther. 2018, 1, e11. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.S.; Ma, Q.; Liu, D.L.; Junghans, R.P. Anti-GD3 chimeric sFv-CD28/T-cell receptor zeta designer T cells for treatment of metastatic melanoma and other neuroectodermal tumors. Clin. Cancer Res. 2010, 16, 2769–2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, P.; Kopecky, C.; Hombach, A.; Zigrino, P.; Mauch, C.; Abken, H. Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 2474–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnasamy, D.; Yu, Z.; Theoret, M.R.; Zhao, Y.; Shrimali, R.K.; Morgan, R.A.; Feldman, S.A.; Restifo, N.P.; Rosenberg, S.A. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J. Clin. Investig. 2010, 120, 3953–3968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargett, T.; Fraser, C.K.; Dotti, G.; Yvon, E.S.; Brown, M.P. BRAF and MEK inhibition variably affect GD2-specific chimeric antigen receptor (CAR) T-cell function in vitro. J. Immunother. 2015, 38, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Kluger, H.M.; DiVito, K.; Berger, A.J.; Halaban, R.; Ariyan, S.; Camp, R.L.; Rimm, D.L. Her2/neu is not a commonly expressed therapeutic target in melanoma—A large cohort tissue microarray study. Melanoma Res. 2004, 14, 207–210. [Google Scholar] [CrossRef]
- Pich, C.; Sarrabayrouse, G.; Teiti, I.; Mariame, B.; Rochaix, P.; Lamant, L.; Favre, G.; Maisongrosse, V.; Tilkin-Mariame, A.F. Melanoma-expressed CD70 is involved in invasion and metastasis. Br. J. Cancer 2016, 114, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef]
- Schlaak, M.; Schmidt, P.; Bangard, C.; Kurschat, P.; Mauch, C.; Abken, H. Regression of metastatic melanoma in a patient by antibody targeting of cancer stem cells. Oncotarget 2012, 3, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Cattaruzza, S.; Nicolosi, P.A.; Braghetta, P.; Pazzaglia, L.; Benassi, M.S.; Picci, P.; Lacrima, K.; Zanocco, D.; Rizzo, E.; Stallcup, W.B.; et al. NG2/CSPG4-collagen type VI interplays putatively involved in the microenvironmental control of tumour engraftment and local expansion. J. Mol. Cell Biol. 2013, 5, 176–193. [Google Scholar] [CrossRef] [PubMed]
- Eisenmann, K.M.; McCarthy, J.B.; Simpson, M.A.; Keely, P.J.; Guan, J.L.; Tachibana, K.; Lim, L.; Manser, E.; Furcht, L.T.; Iida, J. Melanoma chondroitin sulphate proteoglycan regulates cell spreading through Cdc42, Ack-1 and p130cas. Nat. Cell Biol. 1999, 1, 507–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Price, M.A.; Li, G.Y.; Bar-Eli, M.; Salgia, R.; Jagedeeswaran, R.; Carlson, J.H.; Ferrone, S.; Turley, E.A.; McCarthy, J.B. Melanoma proteoglycan modifies gene expression to stimulate tumor cell motility, growth, and epithelial-to-mesenchymal transition. Cancer Res. 2009, 69, 7538–7547. [Google Scholar] [CrossRef] [PubMed]
- Iida, J.; Wilhelmson, K.L.; Ng, J.; Lee, P.; Morrison, C.; Tam, E.; Overall, C.M.; McCarthy, J.B. Cell surface chondroitin sulfate glycosaminoglycan in melanoma: Role in the activation of pro-MMP-2 (pro-gelatinase A). Biochem. J. 2007, 403, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Favoino, E.; Wang, Y.; Ma, Y.; Deng, X.; Wang, X. The CSPG4-specific monoclonal antibody enhances and prolongs the effects of the BRAF inhibitor in melanoma cells. Immunol. Res. 2011, 50, 294–302. [Google Scholar] [CrossRef]
- Dorrie, J.; Babalija, L.; Hoyer, S.; Gerer, K.F.; Schuler, G.; Heinzerling, L.; Schaft, N. BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy. Int. J. Mol. Sci. 2018, 19, 289. [Google Scholar] [CrossRef] [Green Version]
- Beard, R.E.; Abate-Daga, D.; Rosati, S.F.; Zheng, Z.; Wunderlich, J.R.; Rosenberg, S.A.; Morgan, R.A. Gene expression profiling using nanostring digital RNA counting to identify potential target antigens for melanoma immunotherapy. Clin. Cancer Res. 2013, 19, 4941–4950. [Google Scholar] [CrossRef]
- Beard, R.E.; Zheng, Z.; Lagisetty, K.H.; Burns, W.R.; Tran, E.; Hewitt, S.M.; Abate-Daga, D.; Rosati, S.F.; Fine, H.A.; Ferrone, S.; et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J. Immunother. Cancer 2014, 2, 25. [Google Scholar] [CrossRef] [Green Version]
- Geldres, C.; Savoldo, B.; Hoyos, V.; Caruana, I.; Zhang, M.; Yvon, E.; Del, V.M.; Creighton, C.J.; Ittmann, M.; Ferrone, S.; et al. T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin. Cancer Res. 2014, 20, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Schlingemann, R.O.; Rietveld, F.J.; de Waal, R.M.; Ferrone, S.; Ruiter, D.J. Expression of the high molecular weight melanoma-associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am. J. Pathol. 1990, 136, 1393–1405. [Google Scholar]
- Krug, C.; Birkholz, K.; Paulus, A.; Schwenkert, M.; Schmidt, P.; Hoffmann, N.; Hombach, A.; Fey, G.; Abken, H.; Schuler, G.; et al. Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol. Immunother. 2015, 64, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Wiesinger, M.; Marz, J.; Kummer, M.; Schuler, G.; Dorrie, J.; Schuler-Thurner, B.; Schaft, N. Clinical-Scale Production of CAR-T Cells for the Treatment of Melanoma Patients by mRNA Transfection of a CSPG4-Specific CAR under Full GMP Compliance. Cancers 2019, 11, 1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015, 350, aab4077. [Google Scholar] [CrossRef] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, M. The UniCAR system: A modular CAR T cell approach to improve the safety of CAR T cells. Immunol. Lett. 2019, 211, 13–22. [Google Scholar] [CrossRef]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Holzinger, A.; Abken, H. CAR T Cells: A Snapshot on the Growing Options to Design a CAR. Hemasphere 2019, 3, e172. [Google Scholar] [CrossRef]
- Falvo, E.; Tremante, E.; Fraioli, R.; Leonetti, C.; Zamparelli, C.; Boffi, A.; Morea, V.; Ceci, P.; Giacomini, P. Antibody-drug conjugates: Targeting melanoma with cisplatin encapsulated in protein-cage nanoparticles based on human ferritin. Nanoscale 2013, 5, 12278–12285. [Google Scholar] [CrossRef] [Green Version]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bair, S.M.; Porter, D.L. Accelerating chimeric antigen receptor therapy in chronic lymphocytic leukemia: The development and challenges of chimeric antigen receptor T-cell therapy for chronic lymphocytic leukemia. Am. J. Hematol. 2019, 94, S10–S17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Terakura, S.; Martens, A.C.; van, M.T.; Uchiyama, S.; Imai, M.; Sakemura, R.; Goto, T.; Hanajiri, R.; Imahashi, N.; et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 zeta chimeric antigen receptor-modified effector CD8+ T cells. J. Immunol. 2015, 194, 911–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano Attianese, G.M.; Marin, V.; Hoyos, V.; Savoldo, B.; Pizzitola, I.; Tettamanti, S.; Agostoni, V.; Parma, M.; Ponzoni, M.; Bertilaccio, M.T.; et al. In vitro and in vivo model of a novel immunotherapy approach for chronic lymphocytic leukemia by anti-CD23 chimeric antigen receptor. Blood 2011, 117, 4736–4745. [Google Scholar] [CrossRef]
- Hudecek, M.; Schmitt, T.M.; Baskar, S.; Lupo-Stanghellini, M.T.; Nishida, T.; Yamamoto, T.N.; Bleakley, M.; Turtle, C.J.; Chang, W.C.; Greisman, H.A.; et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood 2010, 116, 4532–4541. [Google Scholar] [CrossRef] [Green Version]
- Faitschuk, E.; Hombach, A.A.; Frenzel, L.P.; Wendtner, C.M.; Abken, H. Chimeric antigen receptor T cells targeting Fc mu receptor selectively eliminate CLL cells while sparing healthy B cells. Blood 2016, 128, 1711–1722. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [Green Version]
- Van Bruggen, J.A.C.; Martens, A.W.J.; Fraietta, J.A.; Hofland, T.; Tonino, S.H.; Eldering, E.; Levin, M.D.; Siska, P.J.; Endstra, S.; Rathmell, J.C.; et al. Chronic lymphocytic leukemia cells impair mitochondrial fitness in CD8(+) T cells and impede CAR T-cell efficacy. Blood 2019, 134, 44–58. [Google Scholar] [CrossRef]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Honemann, D.; et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef] [Green Version]
- Cummins, K.D.; Gill, S. Chimeric antigen receptor T-cell therapy for acute myeloid leukemia: How close to reality? Haematologica 2019, 104, 1302–1308. [Google Scholar] [CrossRef] [Green Version]
- Haubner, S.; Perna, F.; Kohnke, T.; Schmidt, C.; Berman, S.; Augsberger, C.; Schnorfeil, F.M.; Krupka, C.; Lichtenegger, F.S.; Liu, X.; et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia 2019, 33, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3(-)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Tasian, S.K.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Kenderian, S.S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Aikawa, V.; Morrissette, J.J.; Scholler, J.; Song, D.; Porter, D.L.; Carroll, M.; et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015, 29, 1637–1647. [Google Scholar] [CrossRef]
- Kim, M.Y.; Yu, K.R.; Kenderian, S.S.; Ruella, M.; Chen, S.; Shin, T.H.; Aljanahi, A.A.; Schreeder, D.; Klichinsky, M.; Shestova, O.; et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell 2018, 173, 1439–1453. [Google Scholar] [CrossRef] [Green Version]
- Smith, F.O.; Rauch, C.; Williams, D.E.; March, C.J.; Arthur, D.; Hilden, J.; Lampkin, B.C.; Buckley, J.D.; Buckley, C.V.; Woods, W.G.; et al. The human homologue of rat NG2, a chondroitin sulfate proteoglycan, is not expressed on the cell surface of normal hematopoietic cells but is expressed by acute myeloid leukemia blasts from poor-prognosis patients with abnormalities of chromosome band 11q23. Blood 1996, 87, 1123–1133. [Google Scholar]
- Harrer, D.C.; Schuler, G.; Dorrie, J.; Schaft, N. CSPG4-Specific CAR T Cells for High-Risk Childhood B Cell Precursor Leukemia. Int. J. Mol. Sci. 2019, 20, 2764. [Google Scholar] [CrossRef] [Green Version]
- Perna, F.; Sadelain, M. Myeloid leukemia switch as immune escape from CD19 chimeric antigen receptor (CAR) therapy. Transl. Cancer Res. 2016, 5, S221–S225. [Google Scholar] [CrossRef] [Green Version]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Prieto, C.; Lopez-Millan, B.; Roca-Ho, H.; Stam, R.W.; Romero-Moya, D.; Rodriguez-Baena, F.J.; Sanjuan-Pla, A.; Ayllon, V.; Ramirez, M.; Bardini, M.; et al. NG2 antigen is involved in leukemia invasiveness and central nervous system infiltration in MLL-rearranged infant B-ALL. Leukemia 2018, 32, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Millan, B.; Sanchez-Martinez, D.; Roca-Ho, H.; Gutierrez-Aguera, F.; Molina, O.; Diaz, d.l.G.; Torres-Ruiz, R.; Fuster, J.L.; Ballerini, P.; Suessbier, U.; et al. NG2 antigen is a therapeutic target for MLL-rearranged B-cell acute lymphoblastic leukemia. Leukemia 2019, 33, 1557–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Starr, R.; Aguilar, B.; Shami, A.F.; Martinez, C.; D’Apuzzo, M.; Barish, M.E.; Forman, S.J.; Jensen, M.C. Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin. Cancer Res. 2012, 18, 2199–2209. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef] [Green Version]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]
- Vitale, D.; Kumar, K.S.; Greve, B.; Jang, B.; Oh, E.S.; Alaniz, L.; Gotte, M. Proteoglycans and glycosaminoglycans as regulators of cancer stem cell function and therapeutic resistance. FEBS J. 2019, 286, 2870–2882. [Google Scholar] [CrossRef] [Green Version]
- Chekenya, M.; Enger, P.O.; Thorsen, F.; Tysnes, B.B.; Al-Sarraj, S.; Read, T.A.; Furmanek, T.; Mahesparan, R.; Levine, J.M.; Butt, A.M.; et al. The glial precursor proteoglycan, NG2, is expressed on tumour neovasculature by vascular pericytes in human malignant brain tumours. Neuropathol. Appl. Neurobiol. 2002, 28, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Chekenya, M.; Hjelstuen, M.; Enger, P.O.; Thorsen, F.; Jacob, A.L.; Probst, B.; Haraldseth, O.; Pilkington, G.; Butt, A.; Levine, J.M.; et al. NG2 proteoglycan promotes angiogenesis-dependent tumor growth in CNS by sequestering angiostatin. FASEB J. 2002, 16, 586–588. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, Y.C.; Zheng, Y.Y.; Xu, J.; Zhang, Y.; Liu, W.L.; Li, Z.Y.; Huang, G.D.; Li, W.P. Furanodienone overcomes temozolomide resistance in glioblastoma through the downregulation of CSPG4-Akt-ERK signalling by inhibiting EGR1-dependent transcription. Phytother. Res. 2019, 33, 1736–1747. [Google Scholar] [CrossRef] [PubMed]
- Pellegatta, S.; Savoldo, B.; Di, I.N.; Corbetta, C.; Chen, Y.; Patane, M.; Sun, C.; Pollo, B.; Ferrone, S.; DiMeco, F.; et al. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci. Transl. Med. 2018, 10, eaao2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svendsen, A.; Verhoeff, J.J.; Immervoll, H.; Brogger, J.C.; Kmiecik, J.; Poli, A.; Netland, I.A.; Prestegarden, L.; Planaguma, J.; Torsvik, A.; et al. Expression of the progenitor marker NG2/CSPG4 predicts poor survival and resistance to ionising radiation in glioblastoma. Acta Neuropathol. 2011, 122, 495–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef]
- Walsh, E.M.; Keane, M.M.; Wink, D.A.; Callagy, G.; Glynn, S.A. Review of Triple Negative Breast Cancer and the Impact of Inducible Nitric Oxide Synthase on Tumor Biology and Patient Outcomes. Crit. Rev. Oncog. 2016, 21, 333–351. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdanifar, M.; Roy, L.D.; Whilding, L.M.; Gavrill, A.; Maher, J.; Mukherjee, P. CAR T Cells Targeting the Tumor MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth. Front. Immunol. 2019, 10, 1149. [Google Scholar] [CrossRef] [Green Version]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef] [Green Version]
- Tchou, J.; Wang, L.C.; Selven, B.; Zhang, H.; Conejo-Garcia, J.; Borghaei, H.; Kalos, M.; Vondeheide, R.H.; Albelda, S.M.; June, C.H.; et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res. Treat. 2012, 133, 799–804. [Google Scholar] [CrossRef] [Green Version]
- Tozbikian, G.; Brogi, E.; Kadota, K.; Catalano, J.; Akram, M.; Patil, S.; Ho, A.Y.; Reis-Filho, J.S.; Weigelt, B.; Norton, L.; et al. Mesothelin expression in triple negative breast carcinomas correlates significantly with basal-like phenotype, distant metastases and decreased survival. PLoS ONE 2014, 9, e114900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Xie, W.; Song, D.G.; Powell, D.J., Jr. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated NKG2D CAR T cells. J. Hematol. Oncol. 2018, 11, 92. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Sun, H.; Zhang, A.; Wu, X.; Li, Y.; Liu, J.; Duan, Y.; Xiao, F.; Wang, H.; Lv, M.; et al. A novel AXL chimeric antigen receptor endows T cells with anti-tumor effects against triple negative breast cancers. Cell. Immunol. 2018, 331, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Byrd, T.T.; Fousek, K.; Pignata, A.; Szot, C.; Samaha, H.; Seaman, S.; Dobrolecki, L.; Salsman, V.S.; Oo, H.Z.; Bielamowicz, K.; et al. TEM8/ANTXR1-Specific CAR T Cells as a Targeted Therapy for Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [Green Version]
- Wallstabe, L.; Gottlich, C.; Nelke, L.C.; Kuhnemundt, J.; Schwarz, T.; Nerreter, T.; Einsele, H.; Walles, H.; Dandekar, G.; Nietzer, S.L.; et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight 2019, 4, 126345. [Google Scholar] [CrossRef] [Green Version]
- Specht, J.M.; Lee, S.; Turtle, C.J.; Berger, C.; Baladrishnan, A.; Srivastava, S.; Voillet, V.; Veatch, J.; Gooley, T.; Mullane, E.; et al. Abstract CT131: A phase I study of adoptive immunotherapy for advanced ROR1+ malignancies with defined subsets of autologous T cells expressing a ROR1-specific chimeric antigen receptor (ROR1-CAR). Cancer Res. 2018, 78. [Google Scholar] [CrossRef]
- Wang, X.; Osada, T.; Wang, Y.; Yu, L.; Sakakura, K.; Katayama, A.; McCarthy, J.B.; Brufsky, A.; Chivukula, M.; Khoury, T.; et al. CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J. Natl. Cancer Inst. 2010, 102, 1496–1512. [Google Scholar] [CrossRef] [Green Version]
- Hsu, N.C.; Nien, P.Y.; Yokoyama, K.K.; Chu, P.Y.; Hou, M.F. High chondroitin sulfate proteoglycan 4 expression correlates with poor outcome in patients with breast cancer. Biochem. Biophys. Res. Commun. 2013, 441, 514–518. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Kim, H.M.; Koo, J.S. Differential Expression of Cancer-Associated Fibroblast-Related Proteins in Ductal Carcinoma in situ According to Molecular Subtype and Stromal Histology. Pathobiology 2018, 85, 311–321. [Google Scholar] [CrossRef]
- Welte, G.; Alt, E.; Devarajan, E.; Krishnappa, S.; Jotzu, C.; Song, Y.H. Interleukin-8 derived from local tissue-resident stromal cells promotes tumor cell invasion. Mol. Carcinog. 2012, 51, 861–868. [Google Scholar] [CrossRef]
- Gibby, K.; You, W.K.; Kadoya, K.; Helgadottir, H.; Young, L.J.; Ellies, L.G.; Chang, Y.; Cardiff, R.D.; Stallcup, W.B. Early vascular deficits are correlated with delayed mammary tumorigenesis in the MMTV-PyMT transgenic mouse following genetic ablation of the NG2 proteoglycan. Breast Cancer Res. 2012, 14, R67. [Google Scholar] [CrossRef] [Green Version]
- Kwei, K.A.; Kung, Y.; Salari, K.; Holcomb, I.N.; Pollack, J.R. Genomic instability in breast cancer: Pathogenesis and clinical implications. Mol. Oncol. 2010, 4, 255–266. [Google Scholar] [CrossRef]
- You, W.K.; Yotsumoto, F.; Sakimura, K.; Adams, R.H.; Stallcup, W.B. NG2 proteoglycan promotes tumor vascularization via integrin-dependent effects on pericyte function. Angiogenesis 2014, 17, 61–76. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrer, D.C.; Dörrie, J.; Schaft, N. CSPG4 as Target for CAR-T-Cell Therapy of Various Tumor Entities–Merits and Challenges. Int. J. Mol. Sci. 2019, 20, 5942. https://doi.org/10.3390/ijms20235942
Harrer DC, Dörrie J, Schaft N. CSPG4 as Target for CAR-T-Cell Therapy of Various Tumor Entities–Merits and Challenges. International Journal of Molecular Sciences. 2019; 20(23):5942. https://doi.org/10.3390/ijms20235942
Chicago/Turabian StyleHarrer, Dennis C., Jan Dörrie, and Niels Schaft. 2019. "CSPG4 as Target for CAR-T-Cell Therapy of Various Tumor Entities–Merits and Challenges" International Journal of Molecular Sciences 20, no. 23: 5942. https://doi.org/10.3390/ijms20235942
APA StyleHarrer, D. C., Dörrie, J., & Schaft, N. (2019). CSPG4 as Target for CAR-T-Cell Therapy of Various Tumor Entities–Merits and Challenges. International Journal of Molecular Sciences, 20(23), 5942. https://doi.org/10.3390/ijms20235942