CGRP Signaling via CALCRL Increases Chemotherapy Resistance and Stem Cell Properties in Acute Myeloid Leukemia

 , , ,
, , ,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
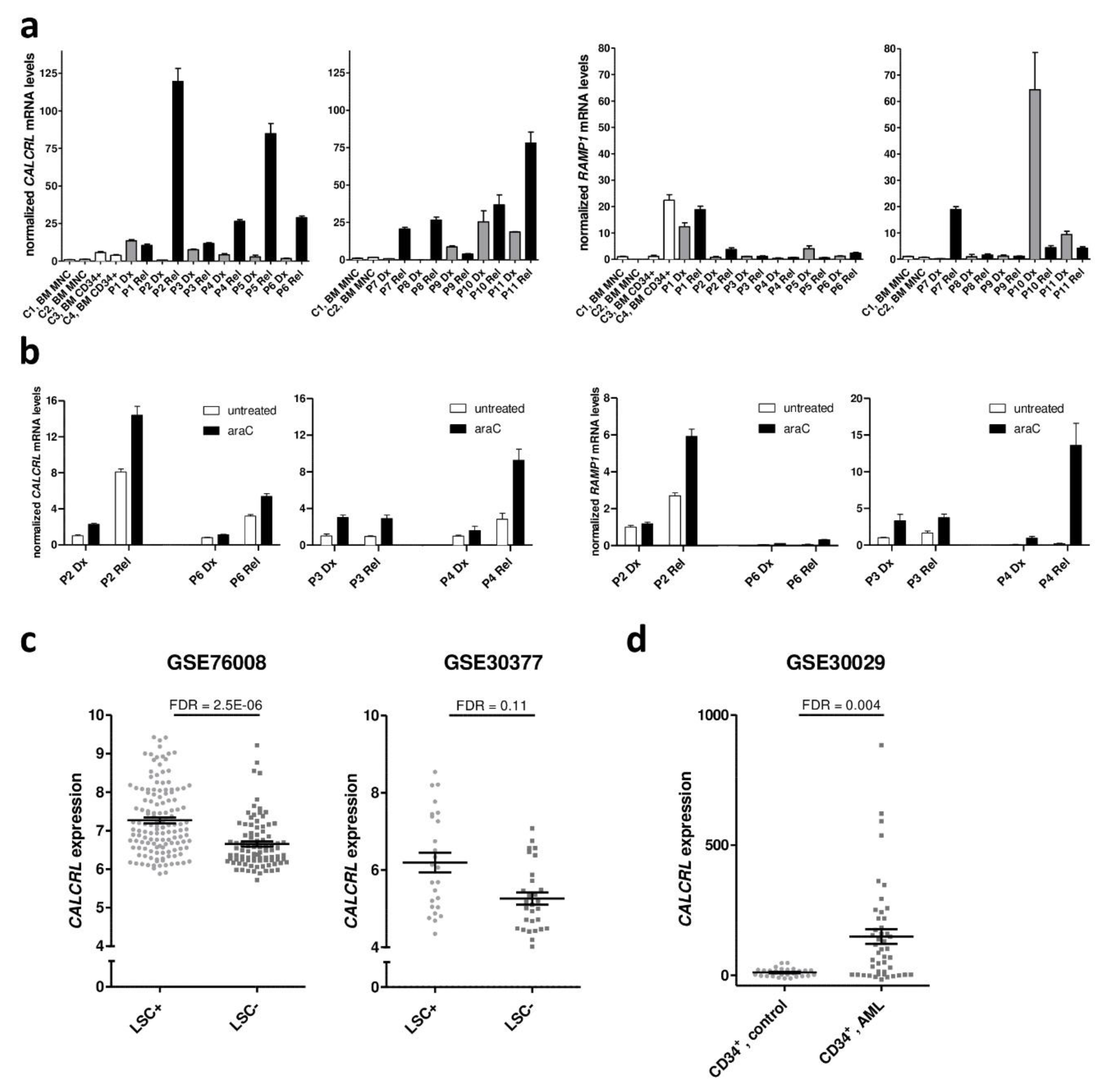
2.1. Expression of CALCRL in Primary Samples from Patients with AML is Associated with Increased Resistance to Chemotherapy
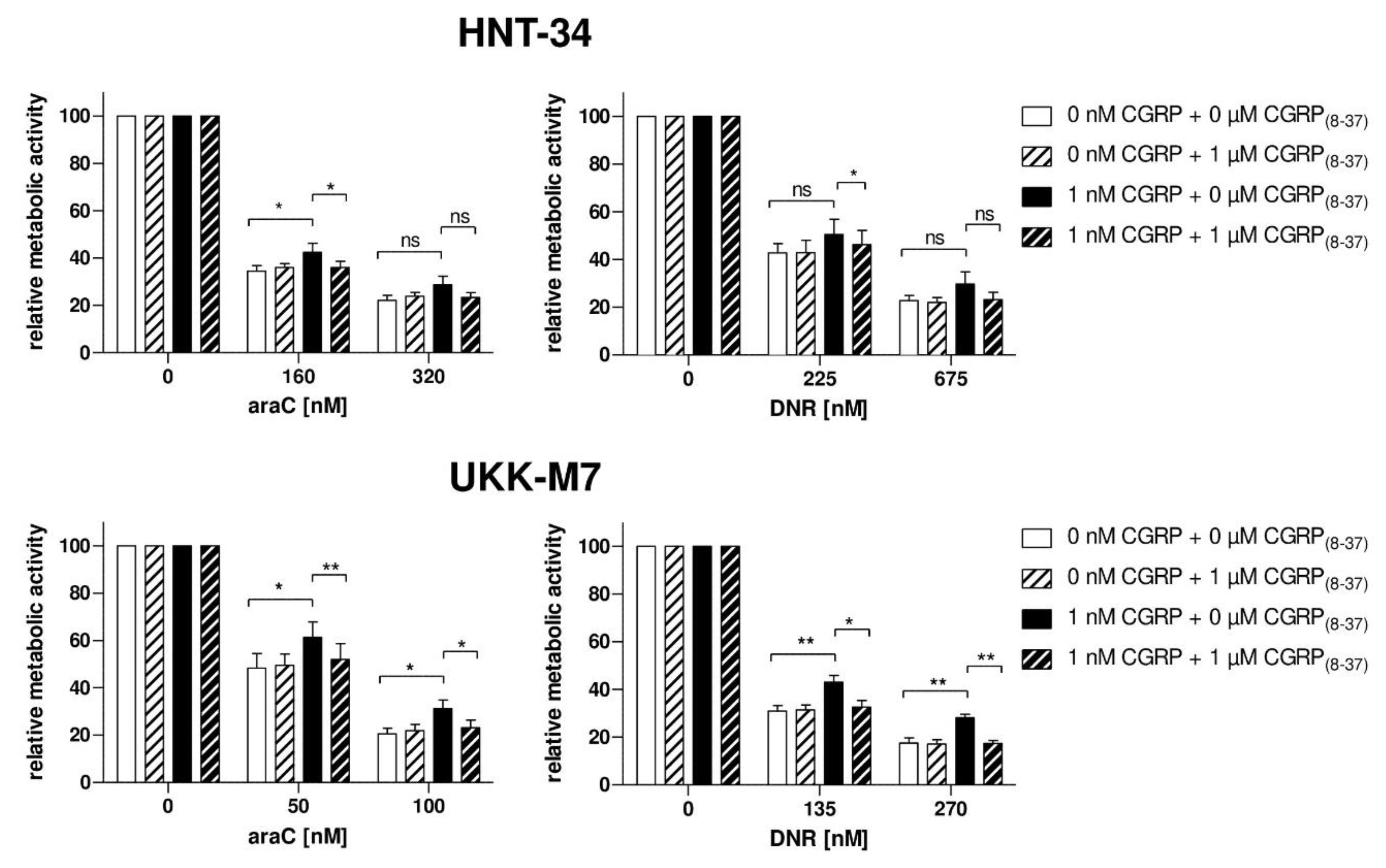
2.2. CGRP Increases the Resistance of Human AML Cell Lines to Drugs Used in the Treatment of AML
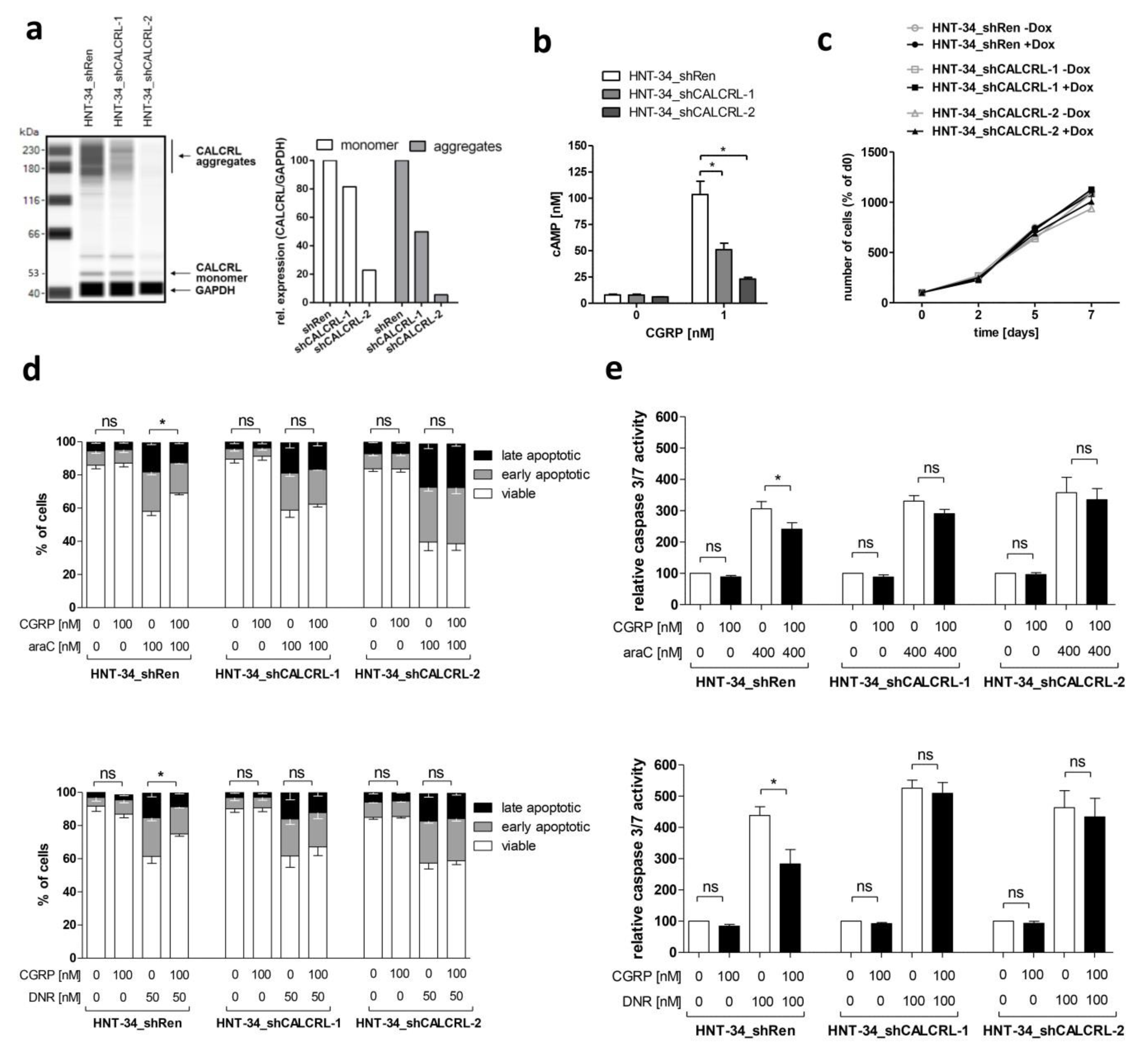
2.3. Genetic Inhibition of CALCRL Counteracts the CGRP-Induced Increase in Chemotherapy Resistance
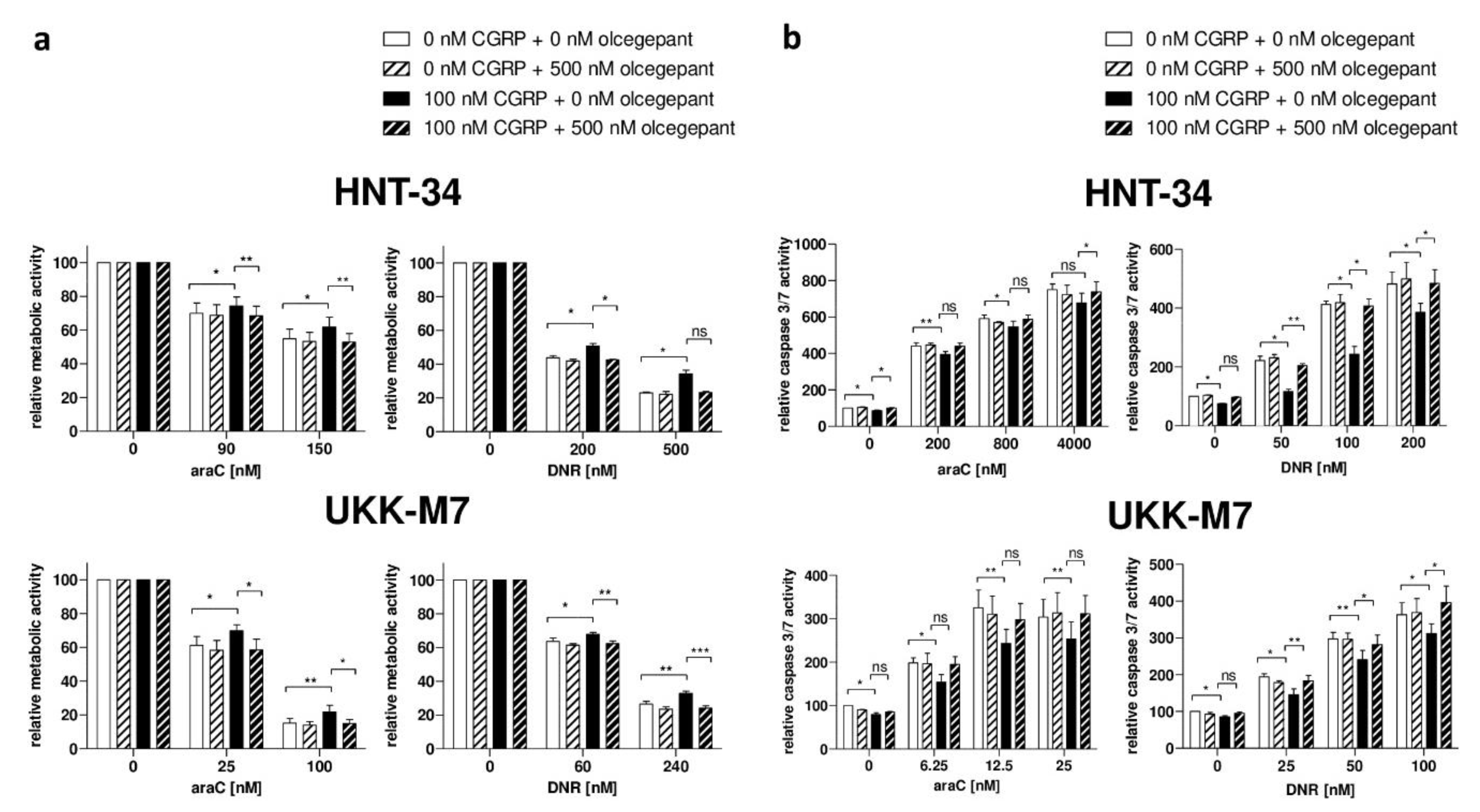
2.4. Pharmacological Inhibition of the CGRP Receptor Counteracts the CGRP-Induced Increase in Chemotherapy Resistance in Human AML Cell Lines and Primary Samples
2.5. In Vivo Treatment with the CGRP Antagonist Olcegepant Increases Differentiation and Decreases Leukemic Burden and Stem Cell Properties in a Mouse Model of AML
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Patient Samples and Healthy Controls
4.3. Establishment of a New AML Cell Line, UKK-M7
4.4. Cell Culture and Retroviral Transductions
4.5. RNA Isolation, Reverse Transcription, and qRT-PCR
4.6. Capillary-Based Protein Quantification Analysis (Wes)
4.7. Metabolic Activity and Apoptosis Assays
4.8. Measurement of cAMP Levels
4.9. Mouse Model
4.10. Statistical and Bioinformatics Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADM | adrenomedullin |
| AML | acute myeloid leukemia |
| araC | cytosine arabinoside |
| BM | bone marrow |
| BM MNC | bone marrow mononuclear cell |
| CALCRL | calcitonin receptor-like receptor |
| cAMP | cyclic adenosine monophosphate |
| CGRP | calcitonin gene-related peptide |
| CGRP(8–37) | truncated CGRP peptide missing amino acids 1–7 |
| CMP | common myeloid progenitor |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DNR | daunorubicin |
| FBS | fetal bovine serum |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GMP | granulocyte/monocyte progenitor |
| HSC | hematopoietic stem cell |
| i.p. | intra-peritoneal |
| LC | leukemic cell |
| LSC | leukemic stem cell |
| LSCe | leukemic stem cell enriched |
| LSK | Lin− Sca-1+ Kit+ |
| M-MLV | moloney murine leukemia virus |
| MA9 | MLL-AF9 |
| MNC | mononuclear cell |
| PBS | phosphate-buffered saline |
| PCR | polymerase chain reaction |
| PLT | platelet |
| qRT-PCR | quantitative real time polymerase chain reaction |
| RAMP | receptor activity modifying protein |
| RBC | red blood cell |
| SD | standard deviation |
| SEM | standard error of the mean |
| shRNA | short hairpin RNA |
| TCGA | The Cancer Genome Atlas |
| WBC | white blood cell |
| Wes | capillary-based protein quantification analysis |
References
- Russell, F.; King, R.; Smillie, S.; Kodji, X.; Brain, S. Calcitonin gene-related peptide: Physiology and pathophysiology. Physiol. Rev. 2014, 94, 1099–1142. [Google Scholar] [CrossRef] [PubMed]
- McLatchie, L.; Fraser, N.; Main, M.; Wise, A.; Brown, J.; Thompson, N.; Solari, R.; Lee, M.; Foord, S. Ramps regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 1998, 393, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.; Walker, C.; Poyner, D. Adrenomedullin and calcitonin gene-related peptide receptors in endocrine-related cancers: Opportunities and challenges. Endocr. Relat. Cancer 2011, 18, C1–C14. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M.; Hirata, Y. Regulation of cell growth and apoptosis by adrenomedullin. Hypertens. Res. 2003, 26, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Spare, N.; Silberstein, S. Targeting cgrp for the prevention of migraine and cluster headache: A narrative review. Headache 2019, 59, 20–32. [Google Scholar] [CrossRef]
- Dallmayer, M.; Li, J.; Ohmura, S.; Alba Rubio, R.; Baldauf, M.; Holting, T.; Musa, J.; Knott, M.; Stein, S.; Cidre-Aranaz, F.; et al. Targeting the calcb/ramp1 axis inhibits growth of ewing sarcoma. Cell Death. Dis. 2019, 10, 116. [Google Scholar] [CrossRef]
- Hagner, S.; Stahl, U.; Grimm, T.; Sturzl, M.; Lang, R. Expression of calcitonin receptor-like receptor in human vascular tumours. J. Clin. Pathol. 2006, 59, 1104–1107. [Google Scholar] [CrossRef]
- Sueur, S.; Pesant, M.; Rochette, L.; Connat, J. Antiapoptotic effect of calcitonin gene-related peptide on oxidative stress-induced injury in h9c2 cardiomyocytes via the ramp1/crlr complex. J. Mol. Cell Cardiol. 2005, 39, 955–963. [Google Scholar] [CrossRef]
- Liu, C.; Chen, X.; Jin, Y.; Qu, R.; Jiang, P.; Wen, G.; Tang, Z. Effects of calcitonin gene-related peptide on apoptosis of peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin. Exp. Dermatol. 2007, 32, 650–653. [Google Scholar] [CrossRef]
- Mrak, E.; Guidobono, F.; Moro, G.; Fraschini, G.; Rubinacci, A.; Villa, I. Calcitonin gene-related peptide (cgrp) inhibits apoptosis in human osteoblasts by beta-catenin stabilization. J. Cell Physiol. 2010, 225, 701–708. [Google Scholar] [CrossRef]
- Toda, M.; Suzuki, T.; Hosono, K.; Hayashi, I.; Hashiba, S.; Onuma, Y.; Amano, H.; Kurihara, Y.; Kurihara, H.; Okamoto, H.; et al. Neuronal system-dependent facilitation of tumor angiogenesis and tumor growth by calcitonin gene-related peptide. Proc. Natl. Acad. Sci. USA 2008, 105, 13550–13555. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Ramos, F. Acute myeloid leukemia in the older adults. Leuk. Res. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sanford, D.; Ravandi, F. Management of newly diagnosed acute myeloid leukemia in the elderly: Current strategies and future directions. Drugs Aging 2015, 32, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, D.; Greystoke, B.; Somervaille, T. The variety of leukemic stem cells in myeloid malignancy. Oncogene 2013, 33, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Zagozdzon, R.; Golab, J. Cancer stem cells in haematological malignancies. Contemp. Oncol. 2015, 19, A1–A6. [Google Scholar] [CrossRef]
- Valk, P.; Verhaak, R.; Beijen, M.; Erpelinck, C.; Barjesteh van Waalwijk van Doorn-Khosrovani, S.; Boer, J.; Beverloo, H.; Moorhouse, M.; van der Spek, P.; Lowenberg, B.; et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N. Engl. J. Med. 2004, 350, 1617–1628. [Google Scholar] [CrossRef]
- Heuser, M.; Wingen, L.; Steinemann, D.; Cario, G.; von Neuhoff, N.; Tauscher, M.; Bullinger, L.; Krauter, J.; Heil, G.; Dohner, H.; et al. Gene-expression profiles and their association with drug resistance in adult acute myeloid leukemia. Haematologica 2005, 90, 1484–1492. [Google Scholar]
- Metzeler, K.; Hummel, M.; Bloomfield, C.; Spiekermann, K.; Braess, J.; Sauerland, M.; Heinecke, A.; Radmacher, M.; Marcucci, G.; Whitman, S.; et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood 2008, 112, 4193–4201. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar]
- Grimwade, D.; Ivey, A.; Huntly, B. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood 2016, 127, 29–41. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.; Paschka, P.; Roberts, N.; Potter, N.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; Mitchell, A.; Kennedy, J.; Chen, W.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Perl, A. The most novel of the novel agents for acute myeloid leukemia. Curr. Opin. Hematol. 2018, 25, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Bohl, S.; Bullinger, L.; Rucker, F. New targeted agents in acute myeloid leukemia: New hope on the rise. Int. J. Mol. Sci. 2019, 20, 1983. [Google Scholar] [CrossRef]
- Szer, J. The prevalent predicament of relapsed acute myeloid leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 43–48. [Google Scholar] [CrossRef]
- Forman, S.; Rowe, J. The myth of the second remission of acute leukemia in the adult. Blood 2013, 121, 1077–1082. [Google Scholar] [CrossRef]
- Hackl, H.; Astanina, K.; Wieser, R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 51. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.; Larson, D.; Miller, C.; Koboldt, D.; Welch, J.; Ritchey, J.; Young, M.; Lamprecht, T.; McLellan, M.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Lowenberg, B. Acute myeloid leukemia: The challenge of capturing disease variety. Hematol. Am. Soc. Hematol. Educ. Program 2008, 2008, 1–11. [Google Scholar] [CrossRef]
- Ayala, F.; Dewar, R.; Kieran, M.; Kalluri, R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia 2009, 23, 2233–2241. [Google Scholar] [CrossRef]
- Meyer, J.; Wang, J.; Hogan, L.; Yang, J.; Dandekar, S.; Patel, J.; Tang, Z.; Zumbo, P.; Li, S.; Zavadil, J.; et al. Relapse-specific mutations in nt5c2 in childhood acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Tzoneva, G.; Perez-Garcia, A.; Carpenter, Z.; Khiabanian, H.; Tosello, V.; Allegretta, M.; Paietta, E.; Racevskis, J.; Rowe, J.; Tallman, M.; et al. Activating mutations in the nt5c2 nucleotidase gene drive chemotherapy resistance in relapsed all. Nat. Med. 2013, 19, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, H.; Jelinek, J.; Estecio, M.; He, R.; Kondo, K.; Chung, W.; Zhang, L.; Shen, L.; Kantarjian, H.; Bueso-Ramos, C.; et al. Aberrant cpg island methylation in acute myeloid leukemia is accentuated at relapse. Blood 2008, 112, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- Hackl, H.; Steinleitner, K.; Lind, K.; Hofer, S.; Tosic, N.; Pavlovic, S.; Suvajdzic, N.; Sill, H.; Wieser, R. A gene expression profile associated with relapse of cytogenetically normal acute myeloid leukemia is enriched for leukemia stem cell genes. Leuk. Lymphoma 2015, 56, 1126–1128. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.; Vadakekolathu, J.; Tasian, S.; Altmann, H.; Bornhauser, M.; Pockley, A.; Ball, G.; Rutella, S. A parsimonious 3-gene signature predicts clinical outcomes in an acute myeloid leukemia multicohort study. Blood Adv. 2019, 3, 1330–1346. [Google Scholar] [CrossRef] [Green Version]
- Irie, K.; Hara-Irie, F.; Ozawa, H.; Yajima, T. Calcitonin gene-related peptide (cgrp)-containing nerve fibers in bone tissue and their involvement in bone remodeling. Microsc. Res. Tech. 2002, 58, 85–90. [Google Scholar] [CrossRef]
- Mach, D.; Rogers, S.; Sabino, M.; Luger, N.; Schwei, M.; Pomonis, J.; Keyser, C.; Clohisy, D.; Adams, D.; O’Leary, P.; et al. Origins of skeletal pain: Sensory and sympathetic innervation of the mouse femur. Neuroscience 2002, 113, 155–166. [Google Scholar] [CrossRef]
- Harzenetter, M.; Keller, U.; Beer, S.; Riedl, C.; Peschel, C.; Holzmann, B. Regulation and function of the cgrp receptor complex in human granulopoiesis. Exp. Hematol. 2002, 30, 306–312. [Google Scholar] [CrossRef]
- Suekane, A.; Saito, Y.; Nakahata, S.; Ichikawa, T.; Ogoh, H.; Tsujikawa, K.; Morishita, K. Cgrp-crlr/ramp1 signal is important for stress-induced hematopoiesis. Sci. Rep. 2019, 9, 429. [Google Scholar] [CrossRef] [Green Version]
- Broome, C.; Whetton, A.; Miyan, J. Neuropeptide control of bone marrow neutrophil production is mediated by both direct and indirect effects on cfu-gm. Br. J. Haematol. 2000, 108, 140–150. [Google Scholar] [CrossRef]
- Angenendt, L.; Bormann, E.; Pabst, C.; Alla, V.; Gorlich, D.; Braun, L.; Dohlich, K.; Schwoppe, C.; Bohlander, S.; Arteaga, M.; et al. The neuropeptide receptor calcitonin receptor-like (calcrl) is a potential therapeutic target in acute myeloid leukemia. Leukemia 2019, 1. [Google Scholar] [CrossRef] [PubMed]
- Eppert, K.; Takenaka, K.; Lechman, E.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, H.; Woolthuis, C.; Vos, A.; Mulder, A.; van den Berg, E.; Kluin, P.; van der Weide, K.; de Bont, E.; Huls, G.; Vellenga, E.; et al. Gene expression profiling in the leukemic stem cell-enriched cd34+ fraction identifies target genes that predict prognosis in normal karyotype aml. Leukemia 2011, 25, 1825–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.; Wouters, B.; Erpelinck, C.; Abbas, S.; Beverloo, H.; Lugthart, S.; Lowenberg, B.; Delwel, R.; Valk, P. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica 2009, 94, 131–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, T.; Jurinovic, V.; Batcha, A.; Bamopoulos, S.; Rothenberg-Thurley, M.; Ksienzyk, B.; Hartmann, L.; Greif, P.; Phillippou-Massier, J.; Krebs, S.; et al. A 29-gene and cytogenetic score for the prediction of resistance to induction treatment in acute myeloid leukemia. Haematologica 2018, 103, 456–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, M.; Chiu, Y.; Chou, W.; Hou, H.; Tseng, M.; Kuo, Y.; Chen, Y.; Chuang, E.; Tien, H. An mrna expression signature for prognostication in de novo acute myeloid leukemia patients with normal karyotype. Oncotarget 2015, 6, 39098–39110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamaguchi, H.; Suzukawa, K.; Nagata, K.; Yamamoto, K.; Yagasaki, F.; Morishita, K. Establishment of a novel human myeloid leukemia cell line (hnt-34) with t(3;3)(q21;q26), t(9;22)(q34;q11) and the expression of evi1 gene, p210 and p190 bcr/abl chimaeric transcripts from a patient with aml after mds with 3q21q26 syndrome. Br. J. Haematol. 1997, 98, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Hofmann, J.; Burmeister, T.; Groger, D.; Park, T.; Emerenciano, M.; Pombo de Oliveira, M.; Renneville, A.; Villarese, P.; Macintyre, E.; et al. The mll recombinome of acute leukemias in 2013. Leukemia 2013, 27, 2165–2176. [Google Scholar] [CrossRef]
- Krivtsov, A.; Twomey, D.; Feng, Z.; Stubbs, M.; Wang, Y.; Faber, J.; Levine, J.; Wang, J.; Hahn, W.; Gilliland, D.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by mll-af9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef]
- Nguyen, C.; Gluxam, T.; Schlerka, A.; Bauer, K.; Grandits, A.; Hackl, H.; Dovey, O.; Zochbauer-Muller, S.; Cooper, J.; Vassiliou, G.; et al. Socs2 is part of a highly prognostic 4-gene signature in aml and promotes disease aggressiveness. Sci. Rep. 2019, 9, 9139. [Google Scholar] [CrossRef]
- Krivtsov, A.; Figueroa, M.; Sinha, A.; Stubbs, M.; Feng, Z.; Valk, P.; Delwel, R.; Dohner, K.; Bullinger, L.; Kung, A.; et al. Cell of origin determines clinically relevant subtypes of mll-rearranged aml. Leukemia 2013, 27, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Vijay, V.; Miller, R.; Vue, G.; Pezeshkian, M.; Maywood, M.; Ast, A.; Drusbosky, L.; Pompeu, Y.; Salgado, A.; Lipten, S.; et al. Interleukin-8 blockade prevents activated endothelial cell mediated proliferation and chemoresistance of acute myeloid leukemia. Leuk. Res. 2019, 84, 106180. [Google Scholar] [CrossRef] [PubMed]
- Noack, K.; Mahendrarajah, N.; Hennig, D.; Schmidt, L.; Grebien, F.; Hildebrand, D.; Christmann, M.; Kaina, B.; Sellmer, A.; Mahboobi, S.; et al. Analysis of the interplay between all-trans retinoic acid and histone deacetylase inhibitors in leukemic cells. Arch. Toxicol. 2017, 91, 2191–2208. [Google Scholar] [CrossRef] [PubMed]
- Hennig, D.; Muller, S.; Wichmann, C.; Drube, S.; Pietschmann, K.; Pelzl, L.; Grez, M.; Bug, G.; Heinzel, T.; Kramer, O. Antagonism between granulocytic maturation and deacetylase inhibitor-induced apoptosis in acute promyelocytic leukaemia cells. Br. J. Cancer 2015, 112, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Fisser, M.; Rommer, A.; Steinleitner, K.; Heller, G.; Herbst, F.; Wiese, M.; Glimm, H.; Sill, H.; Wieser, R. Induction of the proapoptotic tumor suppressor gene cell adhesion molecule 1 by chemotherapeutic agents is repressed in therapy resistant acute myeloid leukemia. Mol. Carcinog. 2015, 54, 1815–1819. [Google Scholar] [CrossRef] [PubMed]
- Fellmann, C.; Hoffmann, T.; Sridhar, V.; Hopfgartner, B.; Muhar, M.; Roth, M.; Lai, D.; Barbosa, I.; Kwon, J.; Guan, Y.; et al. An optimized microrna backbone for effective single-copy rnai. Cell Rep. 2013, 5, 1704–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Russo, A.; Kuburas, A.; Kaiser, E.; Raddant, A.; Recober, A. A potential preclinical migraine model: Cgrp-sensitized mice. Mol. Cell Pharmacol. 2009, 1, 264–270. [Google Scholar]
- Christensen, S.; Petersen, S.; Kristensen, D.; Olesen, J.; Munro, G. Targeting CGRP via receptor antagonism and antibody neutralisation in two distinct rodent models of migraine-like pain. Cephalalgia 2019, 333102419861726. [Google Scholar] [CrossRef]
- Tfelt-Hansen, P.; Olesen, J. Possible site of action of cgrp antagonists in migraine. Cephalalgia 2011, 31, 748–750. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, H.; Akashi, K. Myeloid lineage commitment from the hematopoietic stem cell. Immunity 2007, 26, 726–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuber, J.; Rappaport, A.; Luo, W.; Wang, E.; Chen, C.; Vaseva, A.; Shi, J.; Weissmueller, S.; Fellmann, C.; Taylor, M.; et al. An integrated approach to dissecting oncogene addiction implicates a myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011, 25, 1628–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CALCRL, Univariable | CALCRL, Multivariable | |||||
|---|---|---|---|---|---|---|
| Accession Number | HR | 95% CI | p-Value | HR | 95% CI | p-Value |
| GSE12417, cohort 1 | 2.13 | 1.38–3.3 | 0.0007 | 1.97 | 1.27–3.07 | 0.002 |
| GSE12417, cohort 2 | 2.5 | 1.4–4.5 | 0.002 | 1.76 | 0.89–3.48 | 0.105 |
| GSE6891, cohort 1 | 2.1 | 1.49–2.91 | 1.7 × 10−5 | 1.58 | 1.1–2.3 | 0.012 |
| GSE6891, cohort 2 | 2.65 | 1.8–3.9 | 6.1 × 10−7 | 2.3 | 1.5–3.4 | 6.4 × 10−5 |
| GSE37642 | 1.78 | 1.34–2.35 | 5.6 × 10−5 | 1.64 | 1.2–2.2 | 0.0007 |
| GSE71014 | 4.38 | 2.24–8.56 | 1.5 × 10−5 | n.a. | ||
| TCGA_LAML | 2.09 | 1.26–3.46 | 0.0042 | 1.63 | 0.91–2.74 | 0.066 |
| Target | Clone | Fluorophor | Company | Dilution |
|---|---|---|---|---|
| Mouse Gr-1 | RB6-8C5 | AF700 | Biolegend | 1:100 |
| Mouse Gr-1 | RB6-8C5 | APC | Biolegend | 1:100 |
| Mouse CD11b | M1/70 | AF700 | Biolegend | 1:100 |
| Mouse CD3 | 17A2 | AF700 | Biolegend | 1:100 |
| Mouse B220 | RA3-6B22 | AF700 | Biolegend | 1:100 |
| Mouse Ter119 | TER119 | AF700 | Biolegend | 1:100 |
| Mouse c-Kit | 2B8 | APC-Cy7 | Biolegend | 1:50 |
| Mouse Sca-1 | D7 | PerCP/Cy5.5 | Biolegend | 1:50 |
| Mouse CD34 | MEC14.7 | PE/Cy5.5 | Biolegend | 1:50 |
| Mouse CD16/CD32 | 93 | PE/Cy7 | eBioscience | 1:50 |
| Mouse Ki-67 | 16A8 | APC | Biolegend | 1:50 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gluexam, T.; Grandits, A.M.; Schlerka, A.; Nguyen, C.H.; Etzler, J.; Finkes, T.; Fuchs, M.; Scheid, C.; Heller, G.; Hackl, H.; et al. CGRP Signaling via CALCRL Increases Chemotherapy Resistance and Stem Cell Properties in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2019, 20, 5826. https://doi.org/10.3390/ijms20235826
Gluexam T, Grandits AM, Schlerka A, Nguyen CH, Etzler J, Finkes T, Fuchs M, Scheid C, Heller G, Hackl H, et al. CGRP Signaling via CALCRL Increases Chemotherapy Resistance and Stem Cell Properties in Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2019; 20(23):5826. https://doi.org/10.3390/ijms20235826
Chicago/Turabian StyleGluexam, Tobias, Alexander M. Grandits, Angela Schlerka, Chi Huu Nguyen, Julia Etzler, Thomas Finkes, Michael Fuchs, Christoph Scheid, Gerwin Heller, Hubert Hackl, and et al. 2019. "CGRP Signaling via CALCRL Increases Chemotherapy Resistance and Stem Cell Properties in Acute Myeloid Leukemia" International Journal of Molecular Sciences 20, no. 23: 5826. https://doi.org/10.3390/ijms20235826
APA StyleGluexam, T., Grandits, A. M., Schlerka, A., Nguyen, C. H., Etzler, J., Finkes, T., Fuchs, M., Scheid, C., Heller, G., Hackl, H., Harrer, N., Sill, H., Koller, E., Stoiber, D., Sommergruber, W., & Wieser, R. (2019). CGRP Signaling via CALCRL Increases Chemotherapy Resistance and Stem Cell Properties in Acute Myeloid Leukemia. International Journal of Molecular Sciences, 20(23), 5826. https://doi.org/10.3390/ijms20235826