New Viral Facets in Oral Diseases: The EBV Paradox


{kind=link}
{kind=link}
Abstract
:1. Introduction
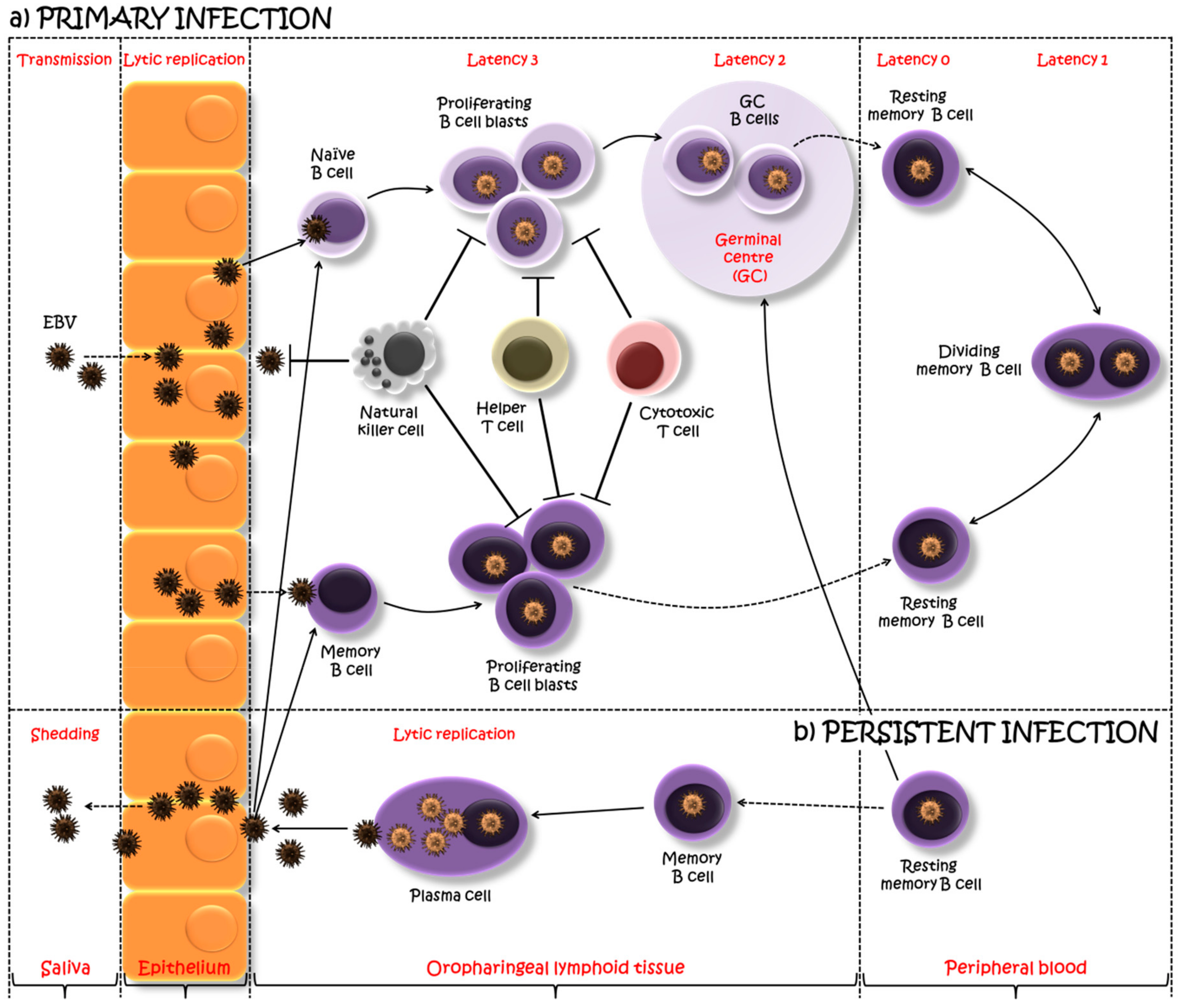
2. EBV Biology
3. Immune Response to EBV
4. The Relationship between EBV and Oral Inflammatory Diseases
4.1. Oral Lichen Planus
4.2. Periodontal Disease
4.3. Sjogren’s Syndrome
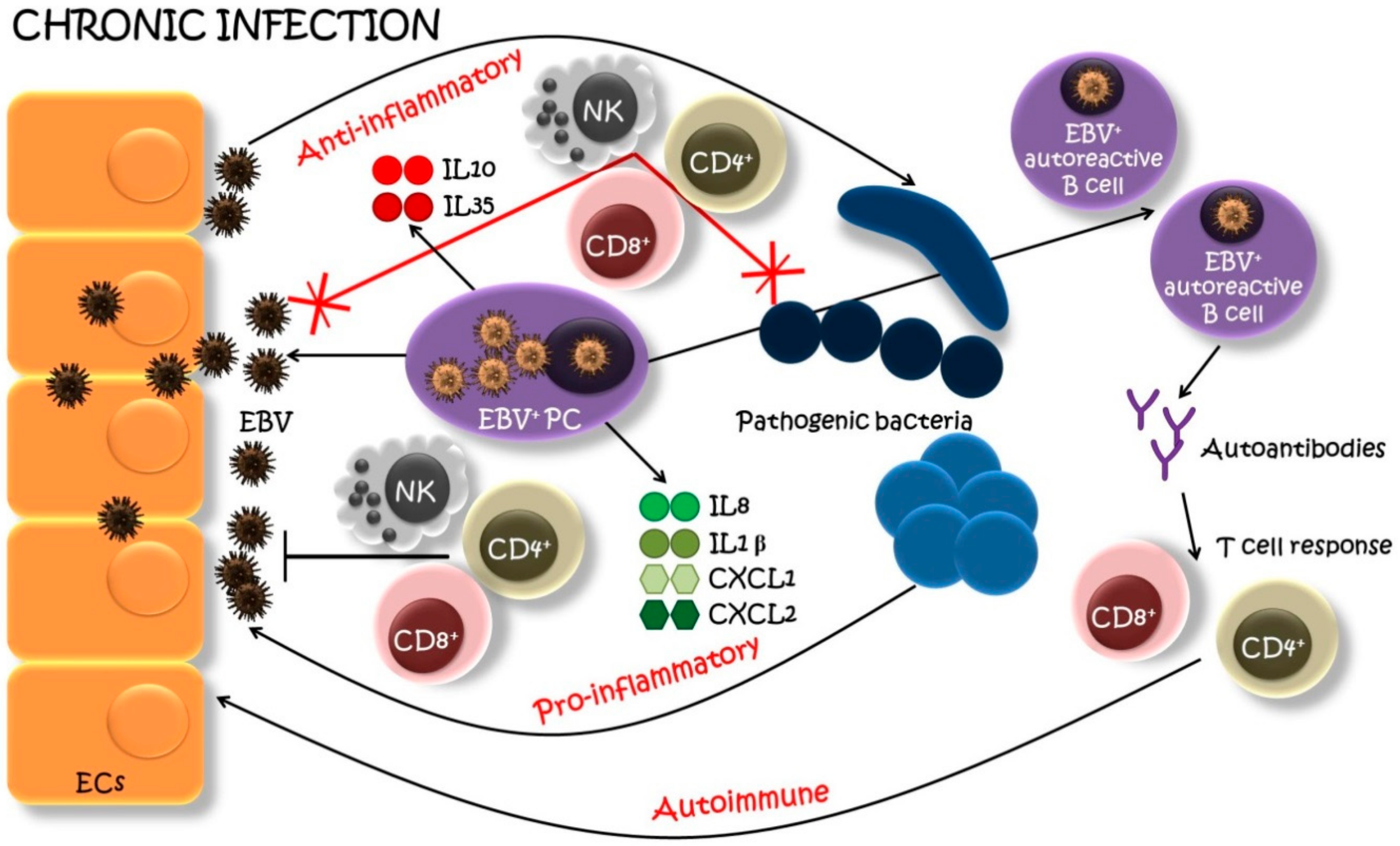
5. Proposed Mechanism of Involvement of EBV-Infected Plasma Cells in the Development and Progression of Oral Inflammatory Diseases
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kilian, M.; Chapple, I.L.C.; Hannig, M.; Marsh, P.D.; Meuric, V.; Pedersen, A.M.L.; Tonetti, M.S.; Wade, W.G.; Zaura, E. The oral microbiome—An update for oral healthcare professionals. Br. Dent. J. 2016, 221, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Slots, J. Human viruses in periodontitis. Periodontol 2000 2010, 53, 89–110. [Google Scholar] [CrossRef] [PubMed]
- Hoare, A.; Marsh, P.D.; Diaz, P.I. Ecological therapeutic opportunities for oral diseases. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Grinde, B.; Olsen, I. The role of viruses in oral disease. J. Oral Microbiol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Abeles, S.R.; Pride, D.T. Molecular bases and role of viruses in the human microbiome. J. Mol. Biol. 2014, 426, 3892–3906. [Google Scholar] [CrossRef] [PubMed]
- Slots, J. Oral viral infections of adults. Periodontol. 2000 2009, 49, 60–86. [Google Scholar] [CrossRef] [PubMed]
- Asai, D.; Nakashima, H. Pathogenic viruses commonly present in the oral cavity and relevant antiviral compounds derived from natural products. Medicines 2018, 5, 120. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, M.M.; Yiu, C.K.; Suter, V.G. Viral causes affecting the oral mucosa. In Management of Dental Emergencies in Children and Adolescents; Neuhaus, K.W., Lussi, A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2019; pp. 217–232. [Google Scholar]
- Clarkson, E.; Mashkoor, F.; Abdulateef, S. Oral viral infections: Diagnosis and management. Dent. Clin. N. Am. 2017, 61, 351–363. [Google Scholar] [CrossRef]
- Abbas, A.A.; Taylor, L.J.; Dothard, M.I.; Leiby, J.S.; Fitzgerald, A.S.; Khatib, L.A.; Collman, R.G.; Bushman, F.D. Redondoviridae, a family of small, circular DNA viruses of the human oro-respiratory tract associated with periodontitis and critical illness. Cell Host Microbe 2019, 25, 719–729.e714. [Google Scholar] [CrossRef]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The immunology of Epstein-Barr virus-induced disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A. EBV persistence—Introducing the virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.D.; Verweij, M.C.; Wiertz, E.J. Herpesviruses and immunity: The art of evasion. Vet. Microbiol. 2010, 143, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.H. Biology and disease associations of Epstein-Barr virus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Stoeger, T.; Adler, H. “Novel” triggers of herpesvirus reactivation and their potential health relevance. Front. Microbiol. 2019, 9, 3207. [Google Scholar] [CrossRef]
- Abbott, R.J.; Pachnio, A.; Pedroza-Pacheco, I.; Leese, A.M.; Begum, J.; Long, H.M.; Croom-Carter, D.; Stacey, A.; Moss, P.A.H.; Hislop, A.D.; et al. Asymptomatic primary infection with epstein-barr virus: Observations on young adult cases. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- CDC. Epstein-Barr Virus and Infectious Mononucleosis. Available online: https://www.cdc.gov/epstein-barr/about-mono.html (accessed on 17 December 2018).
- Hutt-Fletcher, L.M. Epstein-Barr virus entry. J. Virol. 2007, 81, 7825–7832. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Dunmire, S.K.; Grimm, J.M.; Schmeling, D.O.; Balfour, H.H., Jr.; Hogquist, K.A. The incubation period of primary epstein-barr virus infection: Viral dynamics and immunologic events. PLoS Pathog. 2015, 11, e1005286. [Google Scholar] [CrossRef]
- Chesnokova, L.S.; Nishimura, S.L.; Hutt-Fletcher, L.M. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins αvβ6 or αvβ8. Proc. Natl. Acad. Sci. USA 2009, 106, 20464–20469. [Google Scholar] [CrossRef]
- Chesnokova, L.S.; Hutt-Fletcher, L.M. Fusion of Epstein-Barr virus with epithelial cells can be triggered by αvβ5 in addition to αvβ6 and αvβ8, and integrin binding triggers a conformational change in glycoproteins gHgL. J. Virol. 2011, 85, 13214–13223. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; White, B.E.P.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Wang, H.-B.; Zhang, A.; Chen, M.-L.; Fang, Z.-X.; Dong, X.-D.; Li, S.-B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein–Barr virus entry. Nat. Microbiol. 2018, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.; Crawford, D.H. Epstein-Barr virus: The impact of scientific advances on clinical practice. Blood 2006, 107, 862–869. [Google Scholar] [CrossRef]
- Feederle, R.; Kost, M.; Baumann, M.; Janz, A.; Drouet, E.; Hammerschmidt, W.; Delecluse, H.J. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 2000, 19, 3080–3089. [Google Scholar] [CrossRef]
- Murata, T. Regulation of Epstein-Barr virus reactivation from latency. Microbiol. Immunol. 2014, 58, 307–317. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, S.; Sun, R.; Wu, T.; Qian, J. Understanding the interplay between host immunity and Epstein-Barr virus in NPC patients. Emerg. Microbes Infect. 2015, 4, e20. [Google Scholar] [CrossRef]
- Martorelli, D.; Muraro, E.; Merlo, A.; Turrini, R.; Faè, D.A.; Rosato, A.; Dolcetti, R. Exploiting the interplay between innate and adaptive immunity to improve immunotherapeutic strategies for Epstein-Barr-virus-driven disorders. Clin. Dev. Immunol. 2012, 2012, 931952. [Google Scholar] [CrossRef]
- Priatel, J.J.; Chung, B.K.; Tsai, K.; Tan, R. Natural killer T cell strategies to combat Epstein-Barr virus infection. Oncoimmunology 2014, 3, e28329. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, A. Abnormal EBV immune status in oral lichen planus. Oral Dis. 1996, 2, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Adtani, P.; Malathi, N. Epstein-Barr virus and its association with rheumatoid arthritis and oral lichen planus. J. Oral Maxillofac. Pathol. 2015, 19, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Jawanda, M.K. Oral lichen planus: An update on etiology, pathogenesis, clinical presentation, diagnosis and management. Indian J. Dermatol. 2015, 60, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Scully, C.; Beyli, M.; Ferreiro, M.C.; Ficarra, G.; Gill, Y.; Griffiths, M.; Holmstrup, P.; Mutlu, S.; Porter, S.; Wray, D. Update on oral lichen planus: Etiopathogenesis and management. Crit. Rev. Oral Biol. Med. 1998, 9, 86–122. [Google Scholar] [CrossRef]
- Roopashree, M.R.; Gondhalekar, R.V.; Shashikanth, M.C.; George, J.; Thippeswamy, S.H.; Shukla, A. Pathogenesis of oral lichen planus—A review. J. Oral Pathol. Med. 2010, 39, 729–734. [Google Scholar] [CrossRef]
- Sugerman, P.B.; Savage, N.W.; Walsh, L.J.; Zhao, Z.Z.; Zhou, X.J.; Khan, A.; Seymour, G.J.; Bigby, M. The pathogenesis of oral lichen planus. Crit. Rev. Oral Biol. Med. 2002, 13, 350–365. [Google Scholar] [CrossRef] [Green Version]
- Sugerman, P.B.; Savage, N.W. Oral lichen planus: Causes, diagnosis and management. Aust. Dent. J. 2002, 47, 290–297. [Google Scholar] [CrossRef]
- Ismail, S.B.; Kumar, S.K.; Zain, R.B. Oral lichen planus and lichenoid reactions: Etiopathogenesis, diagnosis, management and malignant transformation. J. Oral Sci. 2007, 49, 89–106. [Google Scholar] [CrossRef] [Green Version]
- Walsh, L.J.; Savage, N.W.; Ishii, T.; Seymour, G.J. Immunopathogenesis of oral lichen planus. J. Oral Pathol. Med. 1990, 19, 389–396. [Google Scholar] [CrossRef]
- Cruz, I.; Van den Brule, A.J.; Steenbergen, R.D.; Snijders, P.J.; Meijer, C.J.; Walboomers, J.M.; Snow, G.B.; Van der Waal, I. Prevalence of Epstein-Barr virus in oral squamous cell carcinomas, premalignant lesions and normal mucosa—A study using the polymerase chain reaction. Oral Oncol. 1997, 33, 182–188. [Google Scholar] [CrossRef]
- Sand, L.P.; Jalouli, J.; Larsson, P.A.; Hirsch, J.M. Prevalence of Epstein-Barr virus in oral squamous cell carcinoma, oral lichen planus, and normal oral mucosa. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2002, 93, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Kis, A.; Feher, E.; Gall, T.; Tar, I.; Boda, R.; Toth, E.D.; Mehes, G.; Gergely, L.; Szarka, K. Epstein-Barr virus prevalence in oral squamous cell cancer and in potentially malignant oral disorders in an eastern Hungarian population. Eur. J. Oral Sci. 2009, 117, 536–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shariati, M.; Mokhtari, M.; Masoudifar, A. Association between oral lichen planus and Epstein-Barr virus in Iranian patients. J. Res. Med. Sci. 2018, 23, 24. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, B.; Senguven, B.; Demir, C. Prevalence of herpes simplex, Epstein Barr and human papilloma viruses in oral lichen planus. Med. Oral Patol. Oral Cir. Bucal. 2011, 16, e170–e174. [Google Scholar] [CrossRef] [Green Version]
- Danielsson, K.; Nylander, E.; Sjöström, M.; Ebrahimi, M. Epstein-Barr virus is not detected in mucosal lichen planus. Med. Oral Patol. Oral Cir. Bucal. 2018, 23, e560–e563. [Google Scholar] [CrossRef]
- Raybaud, H.; Olivieri, C.V.; Lupi-Pegurier, L.; Pagnotta, S.; Marsault, R.; Cardot-Leccia, N.; Doglio, A. Epstein-Barr virus-infected plasma cells infiltrate erosive oral lichen planus. J. Dent. Res. 2018. [Google Scholar] [CrossRef]
- Hiepe, F.; Alexander, T.; Voll, R.E. Plasma cells. Z. Rheumatol. 2015, 74, 20–25. [Google Scholar] [CrossRef]
- Fillatreau, S. Regulatory plasma cells. Curr. Opin. Pharmacol. 2015, 23, 1–5. [Google Scholar] [CrossRef]
- Fillatreau, S. Regulatory roles of B cells in infectious diseases. Clin. Exp. Rheumatol. 2016, 34, 1–5. [Google Scholar]
- EFP. Available online: https://www.efp.org/publications/EFP_Dossier_on_Periodontal_Disease_2018.pdf (accessed on 10 September 2018).
- Kinane, D.F.; Stathopoulou, P.G.; Papapanou, P.N. Periodontal diseases. Nat. Rev. Dis. Primers 2017, 3, 17038. [Google Scholar] [CrossRef] [PubMed]
- WHO. Available online: http://www.who.int/oral_health/publications/factsheet/en/ (accessed on 29 August 2019).
- Kim, J.; Amar, S. Periodontal disease and systemic conditions: A bidirectional relationship. Odontology 2006, 94, 10–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, F.Q.; Almeida-da-Silva, C.L.C.; Huynh, B.; Trinh, A.; Liu, J.; Woodward, J.; Asadi, H.; Ojcius, D.M. Association between periodontal pathogens and systemic disease. Biomed. J. 2019, 42, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Michaud, D.S.; Lu, J.; Peacock-Villada, A.Y.; Barber, J.R.; Joshu, C.E.; Prizment, A.E.; Beck, J.D.; Offenbacher, S.; Platz, E.A. Periodontal disease assessed using clinical dental measurements and cancer risk in the ARIC study. J. Natl. Cancer Inst. 2018, 110, 843–854. [Google Scholar] [CrossRef]
- Slots, J. Periodontal herpesviruses: Prevalence, pathogenicity, systemic risk. Periodontol. 2000 2015, 69, 28–45. [Google Scholar] [CrossRef]
- Beader, N.; Ivic-Kardum, M. The role of cytomegalovirus infection in the pathogenesis of periodontal diseases. Acta Clin. Croat. 2011, 50, 61–66. [Google Scholar]
- Slots, J.; Slots, H. Periodontal herpesvirus morbidity and treatment. Periodontol. 2000 2019, 79, 210–220. [Google Scholar] [CrossRef]
- Slots, J. Focal infection of periodontal origin. Periodontol. 2000 2019, 79, 233–235. [Google Scholar] [CrossRef]
- Slots, J. Herpesviruses in periodontal diseases. Periodontol. 2000 2005, 38, 33–62. [Google Scholar] [CrossRef]
- Slots, J. Herpesviral-bacterial synergy in the pathogenesis of human periodontitis. Curr. Opin. Infect. Dis. 2007, 20, 278–283. [Google Scholar] [CrossRef]
- Gao, Z.; Lv, J.; Wang, M. Epstein-Barr virus is associated with periodontal diseases: A meta-analysis based on 21 case-control studies. Medicine 2017, 96, e5980. [Google Scholar] [CrossRef]
- Slots, J. Herpesviral-bacterial interactions in periodontal diseases. Periodontol. 2000 2010, 52, 117–140. [Google Scholar] [CrossRef]
- Imai, K.; Inoue, H.; Tamura, M.; Cueno, M.E.; Inoue, H.; Takeichi, O.; Kusama, K.; Saito, I.; Ochiai, K. The periodontal pathogen Porphyromonas gingivalis induces the Epstein-Barr virus lytic switch transactivator ZEBRA by histone modification. Biochimie 2012, 94, 839–846. [Google Scholar] [CrossRef]
- Makino, K.; Takeichi, O.; Imai, K.; Inoue, H.; Hatori, K.; Himi, K.; Saito, I.; Ochiai, K.; Ogiso, B. Porphyromonas endodontalis reactivates latent Epstein-Barr virus. Int. Endod. J. 2018, 51, 1410–1419. [Google Scholar] [CrossRef]
- Vincent-Bugnas, S.; Vitale, S.; Mouline, C.C.; Khaali, W.; Charbit, Y.; Mahler, P.; Prêcheur, I.; Hofman, P.; Maryanski, J.L.; Doglio, A. EBV infection is common in gingival epithelial cells of the periodontium and worsens during chronic periodontitis. PLoS ONE 2013, 8, e80336. [Google Scholar] [CrossRef] [Green Version]
- Comerford, I.; Bunting, M.; Fenix, K.; Haylock-Jacobs, S.; Litchfield, W.; Harata-Lee, Y.; Turvey, M.; Brazzatti, J.; Gregor, C.; Nguyen, P.; et al. An immune paradox: How can the same chemokine axis regulate both immune tolerance and activation? CCR6/CCL20: A chemokine axis balancing immunological tolerance and inflammation in autoimmune disease. Bioessays 2010, 32, 1067–1076. [Google Scholar] [CrossRef]
- Imai, K.; Ogata, Y.; Ochiai, K. Microbial interaction of periodontopathic bacteria and Epstein-Barr virus and their implication of periodontal diseases. J. Oral Biosci. 2012, 54, 164–168. [Google Scholar] [CrossRef] [Green Version]
- Sandhya, P.; Kurien, B.T.; Danda, D.; Scofield, R.H. Update on pathogenesis of Sjogren’s syndrome. Curr. Rheumatol. Rev. 2017, 13, 5–22. [Google Scholar] [CrossRef]
- Klippel, J.H.; Stone, J.H.; Crofford, L.J.; White, P.H. Primer on the Rheumatic Diseases, 13th ed.; Springer: New York, NY, USA, 2008. [Google Scholar]
- Yamamoto, K. Pathogenesis of Sjogren’s syndrome. Autoimmun. Rev. 2003, 2, 13–18. [Google Scholar] [CrossRef]
- Patel, R.; Shahane, A. The epidemiology of Sjögren’s syndrome. Clin. Epidemiol. 2014, 6, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Kassan, S.S.; Moutsopoulos, H.M. Clinical manifestations and early diagnosis of Sjogren syndrome. Arch. Intern. Med. 2004, 164, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Carrasco, M.; Fuentes-Alexandro, S.; Escarcega, R.O.; Salgado, G.; Riebeling, C.; Cervera, R. Pathophysiology of Sjogren’s syndrome. Arch. Med. Res. 2006, 37, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.I. Sjogren’s syndrome. Lancet 2005, 366, 321–331. [Google Scholar] [CrossRef]
- Fox, R.I.; Saito, I. Sjogren’s syndrome. In Autoimmune Diseases of the Skin: Pathogenesis, Diagnosis, Management; Hertl, M., Ed.; Springer Vienna: Vienna, Austria, 2005; pp. 261–289. [Google Scholar]
- Nocturne, G.; Mariette, X. Advances in understanding the pathogenesis of primary Sjogren’s syndrome. Nat. Rev. Rheumatol. 2013, 9, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Venables, P.J.; Teo, C.G.; Baboonian, C.; Griffin, B.E.; Hughes, R.A. Persistence of Epstein-Barr virus in salivary gland biopsies from healthy individuals and patients with Sjogren’s syndrome. Clin. Exp. Immunol. 1989, 75, 359–364. [Google Scholar]
- Venables, P.J.; Baboonian, C.; Horsfall, A.C.; Halliday, D.; Maini, R.N.; Teo, C.G.; Mageed, R.; Millman, G. The response to Epstein-Barr virus infection in Sjogren’s syndrome. J. Autoimmun. 1989, 2, 439–448. [Google Scholar] [CrossRef]
- DiGiuseppe, J.A.; Wu, T.C.; Corio, R.L. Analysis of Epstein-Barr virus-encoded small RNA 1 expression in benign lymphoepithelial salivary gland lesions. Mod. Pathol. 1994, 7, 555–559. [Google Scholar]
- Fox, R.I.; Pearson, G.; Vaughan, J.H. Detection of Epstein-Barr virus-associated antigens and DNA in salivary gland biopsies from patients with Sjogren’s syndrome. J. Immunol. 1986, 137, 3162–3168. [Google Scholar]
- Saito, I.; Servenius, B.; Compton, T.; Fox, R.I. Detection of Epstein-Barr virus DNA by polymerase chain reaction in blood and tissue biopsies from patients with Sjogren’s syndrome. J. Exp. Med. 1989, 169, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Mariette, X.; Gozlan, J.; Clerc, D.; Bisson, M.; Morinet, F. Detection of Epstein-Barr virus DNA by in situ hybridization and polymerase chain reaction in salivary gland biopsy specimens from patients with Sjogren’s syndrome. Am. J. Med. 1991, 90, 286–294. [Google Scholar] [CrossRef]
- Inoue, N.; Harada, S.; Miyasaka, N.; Oya, A.; Yanagi, K. Analysis of antibody titers to Epstein-Barr virus nuclear antigens in sera of patients with Sjögren’s syndrome and with rheumatoid arthritis. J. Infect. Dis. 1991, 164, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Pflugfelder, S.C.; Crouse, C.A.; Monroy, D.; Yen, M.; Rowe, M.; Atherton, S.S. Epstein-Barr virus and the lacrimal gland pathology of Sjögren’s syndrome. Am. J. Pathol. 1993, 143, 49–64. [Google Scholar] [PubMed]
- Toda, I.; Ono, M.; Fujishima, H.; Tsubota, K. Sjogren’s syndrome (SS) and Epstein-Barr virus (EBV) reactivation. Ocul. Immunol. Inflamm. 1994, 2, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Shimizu, N.; Yoshiyama, H.; Mizugaki, Y.; Shinozaki, F.; Takada, K. Association of Epstein-Barr virus (EBV) with Sjögren’s syndrome: Differential EBV expression between epithelial cells and lymphocytes in salivary glands. Am. J. Pathol. 1996, 149, 1511–1517. [Google Scholar]
- Tateishi, M.; Saito, I.; Yamamoto, K.; Miyasaka, N. Spontaneous production of Epstein-Barr virus by B lymphoblastoid cell lines obtained from patients with Sjogren’s syndrome. Possible involvement of a novel strain of Epstein-Barr virus in disease pathogenesis. Arthritis Rheum. 1993, 36, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Tsubota, K.; Fujishima, H.; Toda, I.; Katagiri, S.; Kawashima, Y.; Saito, I. Increased levels of Epstein-Barr virus DNA in lacrimal glands of Sjogren’s syndrome patients. Acta Ophthalmol. Scand. 1995, 73, 425–430. [Google Scholar] [CrossRef]
- Kivity, S.; Arango, M.T.; Ehrenfeld, M.; Tehori, O.; Shoenfeld, Y.; Anaya, J.-M.; Agmon-Levin, N. Infection and autoimmunity in Sjogren’s syndrome: A clinical study and comprehensive review. J. Autoimmun. 2014, 51, 17–22. [Google Scholar] [CrossRef]
- Pasoto, S.G.; Natalino, R.R.; Chakkour, H.P.; Vdos, S.V.; Bueno, C.; Leon, E.P.; Vendramini, M.B.; Neto, M.L.; Bonfa, E. EBV reactivation serological profile in primary Sjogren’s syndrome: An underlying trigger of active articular involvement? Rheumatol. Int. 2013, 33, 1149–1157. [Google Scholar] [CrossRef]
- Song, G.G.; Kim, J.H.; Seo, Y.H.; Choi, S.J.; Ji, J.D.; Lee, Y.H. Meta-analysis of differentially expressed genes in primary Sjogren’s syndrome by using microarray. Hum. Immunol. 2014, 75, 98–104. [Google Scholar] [CrossRef]
- Croia, C.; Astorri, E.; Murray-Brown, W.; Willis, A.; Brokstad, K.A.; Sutcliffe, N.; Piper, K.; Jonsson, R.; Tappuni, A.R.; Pitzalis, C.; et al. Implication of Epstein-Barr virus infection in disease-specific autoreactive B cell activation in ectopic lymphoid structures of Sjogren’s syndrome. Arthritis Rheumatol. 2014, 66, 2545–2557. [Google Scholar] [CrossRef]
- Bombardieri, M.; Barone, F.; Lucchesi, D.; Nayar, S.; van den Berg, W.B.; Proctor, G.; Buckley, C.D.; Pitzalis, C. Inducible tertiary lymphoid structures, autoimmunity, and exocrine dysfunction in a novel model of salivary gland inflammation in C57BL/6 mice. J. Immunol. 2012, 189, 3767–3776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harley, J.B.; Zoller, E.E. Editorial: What caused all these troubles, anyway? Epstein-Barr virus in Sjogren’s syndrome reevaluated. Arthritis Rheum. 2014, 66, 2328–2330. [Google Scholar] [CrossRef] [PubMed]
- Jing, L.; Kim, S.; Sun, L.; Wang, L.; Mildner, E.; Divaris, K.; Jiao, Y.; Offenbacher, S. IL-37- and IL-35/IL-37-producing plasma cells in chronic periodontitis. J. Dent. Res. 2019, 98, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Mahanonda, R.; Champaiboon, C.; Subbalekha, K.; Sa-Ard-Iam, N.; Rattanathammatada, W.; Thawanaphong, S.; Rerkyen, P.; Yoshimura, F.; Nagano, K.; Lang, N.P.; et al. Human memory B cells in healthy gingiva, gingivitis, and periodontitis. J. Immunol. 2016, 197, 715–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurence, M.; Benito-Leon, J. Epstein-Barr virus and multiple sclerosis: Updating Pender’s hypothesis. Mult. Scler. Relat. Disord. 2017, 16, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Münz, C.; Lünemann, J.D.; Getts, M.T.; Miller, S.D. Antiviral immune responses: Triggers of or triggered by autoimmunity? Nat. Rev. Immunol. 2009, 9, 246–258. [Google Scholar] [CrossRef]
- Tracy, S.I.; Kakalacheva, K.; Lunemann, J.D.; Luzuriaga, K.; Middeldorp, J.; Thorley-Lawson, D.A. Persistence of Epstein-Barr virus in self-reactive memory B cells. J. Virol. 2012, 86, 12330–12340. [Google Scholar] [CrossRef] [Green Version]
- Shannon-Lowe, C.; Rowe, M. Epstein-Barr virus infection of polarized epithelial cells via the basolateral surface by memory B cell-mediated transfer infection. PLoS Pathog. 2011, 7, e1001338. [Google Scholar] [CrossRef] [Green Version]
- Pender, M.P. Infection of autoreactive B lymphocytes with EBV, causing chronic autoimmune diseases. Trends Immunol. 2003, 24, 584–588. [Google Scholar] [CrossRef] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonoyan, L.; Vincent-Bugnas, S.; Olivieri, C.-V.; Doglio, A. New Viral Facets in Oral Diseases: The EBV Paradox. Int. J. Mol. Sci. 2019, 20, 5861. https://doi.org/10.3390/ijms20235861
Tonoyan L, Vincent-Bugnas S, Olivieri C-V, Doglio A. New Viral Facets in Oral Diseases: The EBV Paradox. International Journal of Molecular Sciences. 2019; 20(23):5861. https://doi.org/10.3390/ijms20235861
Chicago/Turabian StyleTonoyan, Lilit, Séverine Vincent-Bugnas, Charles-Vivien Olivieri, and Alain Doglio. 2019. "New Viral Facets in Oral Diseases: The EBV Paradox" International Journal of Molecular Sciences 20, no. 23: 5861. https://doi.org/10.3390/ijms20235861
APA StyleTonoyan, L., Vincent-Bugnas, S., Olivieri, C. -V., & Doglio, A. (2019). New Viral Facets in Oral Diseases: The EBV Paradox. International Journal of Molecular Sciences, 20(23), 5861. https://doi.org/10.3390/ijms20235861