Establishing a System for Functional Characterization of Full-Length cDNAs of Camellia sinensis

Abstract
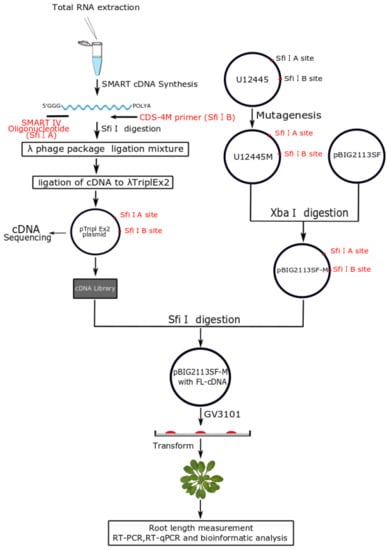
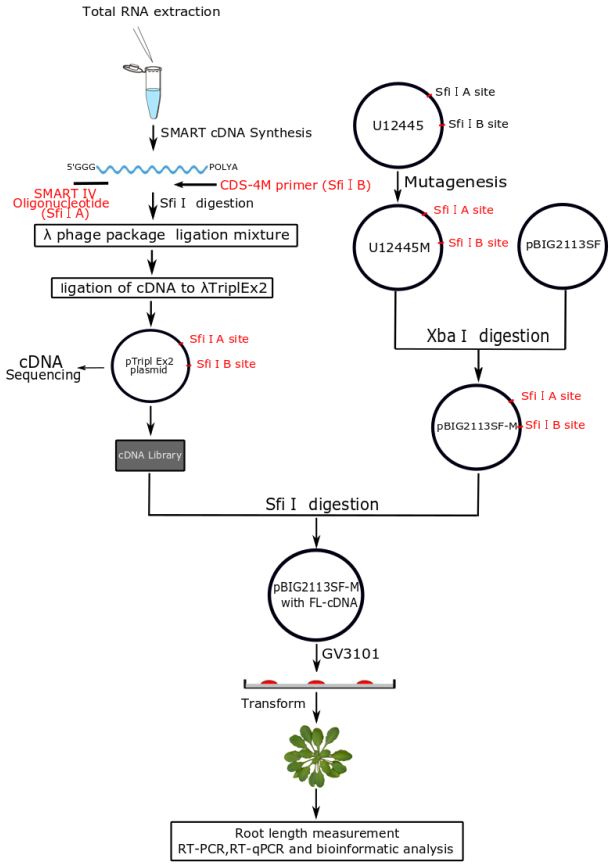
:
1. Introduction
2. Results and Discussion
2.1. Construction of a Normalized Full-Length Enriched cDNA Library of Camellia sinensis
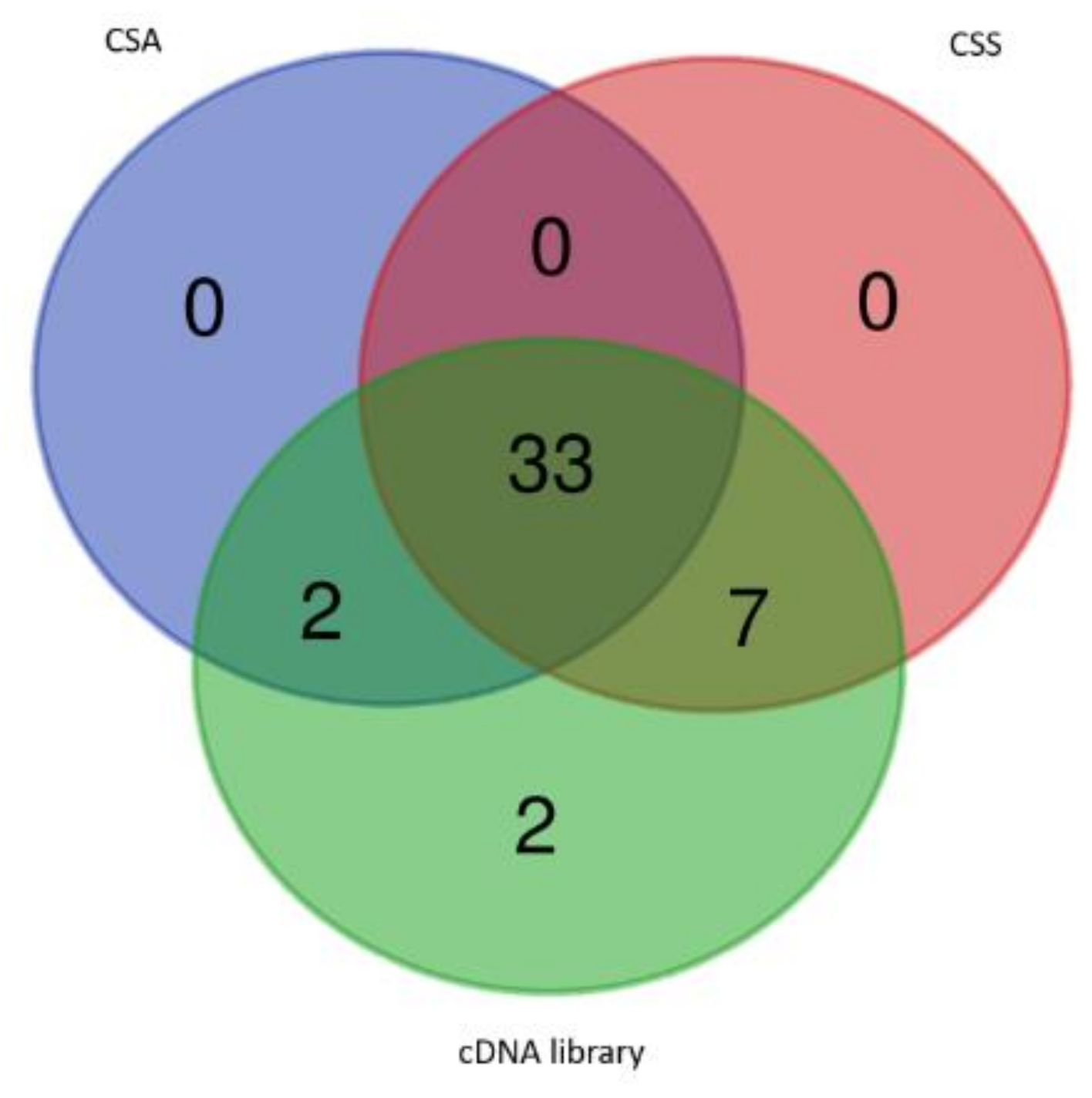
2.2. Comparison with Tea CSS and CSA Genome
2.3. 5′-UTR and 3′-UTR Analysis
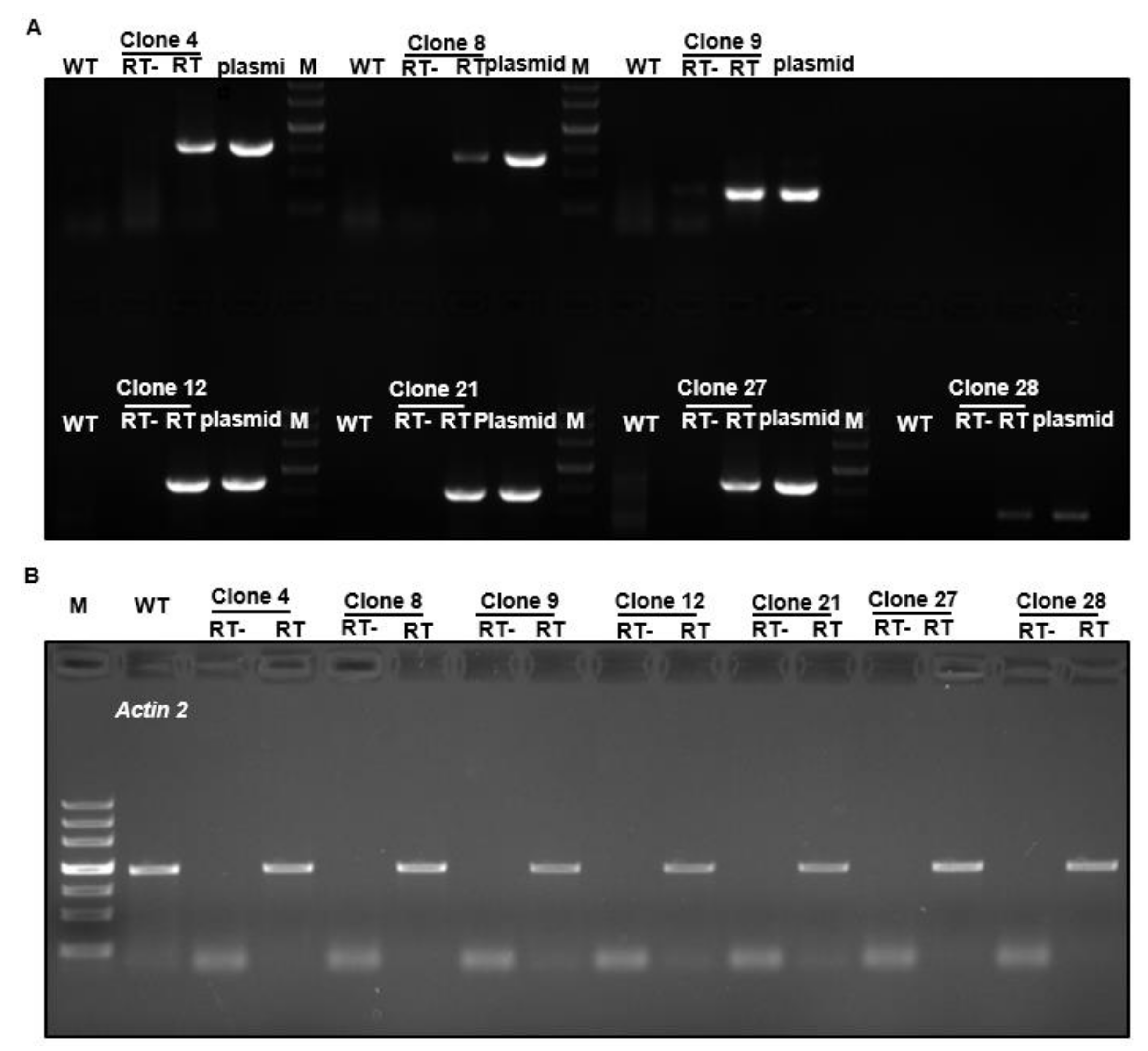
2.4. Ectopic Expression of Tea Genes in Arabidopsis
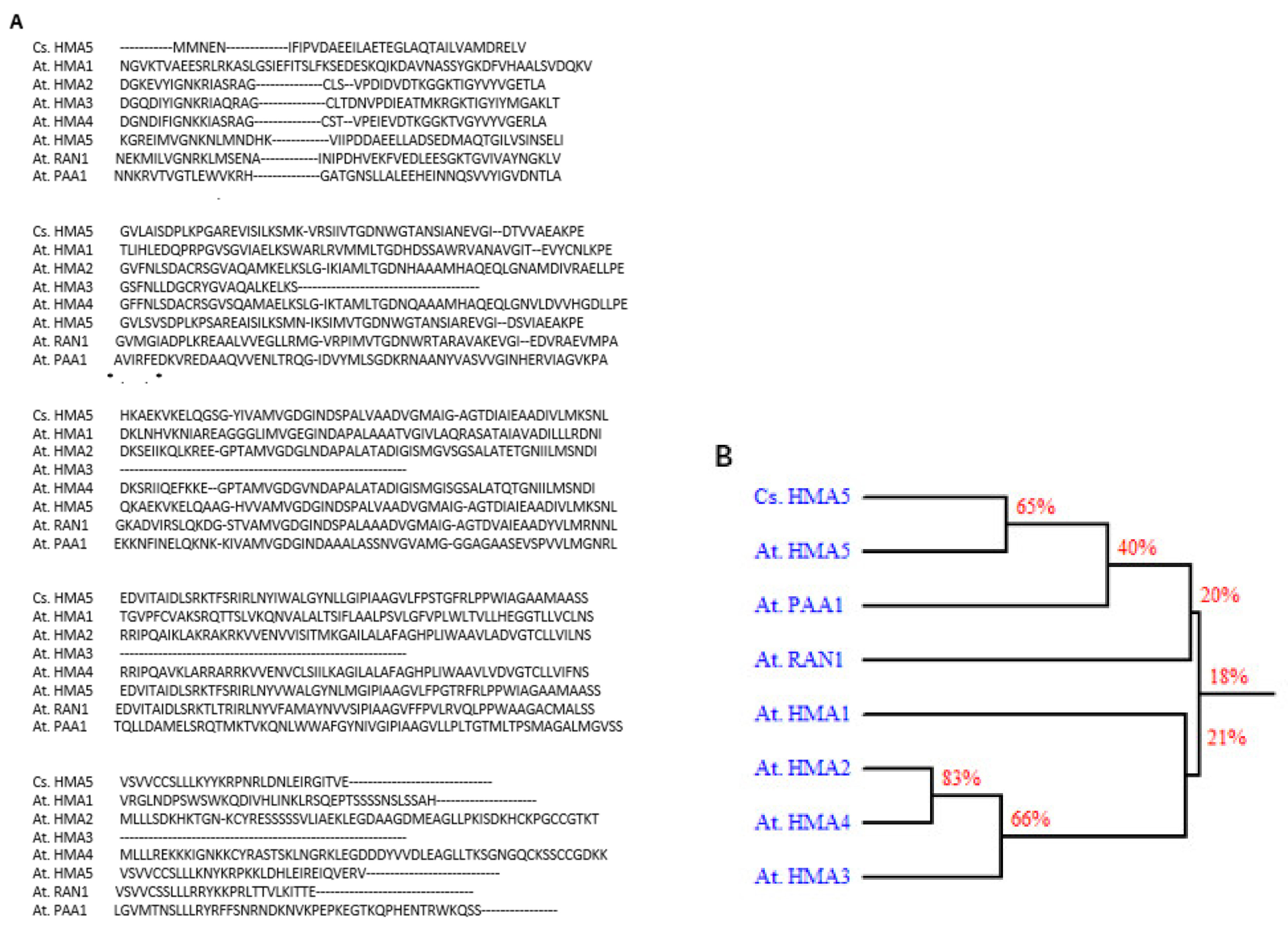
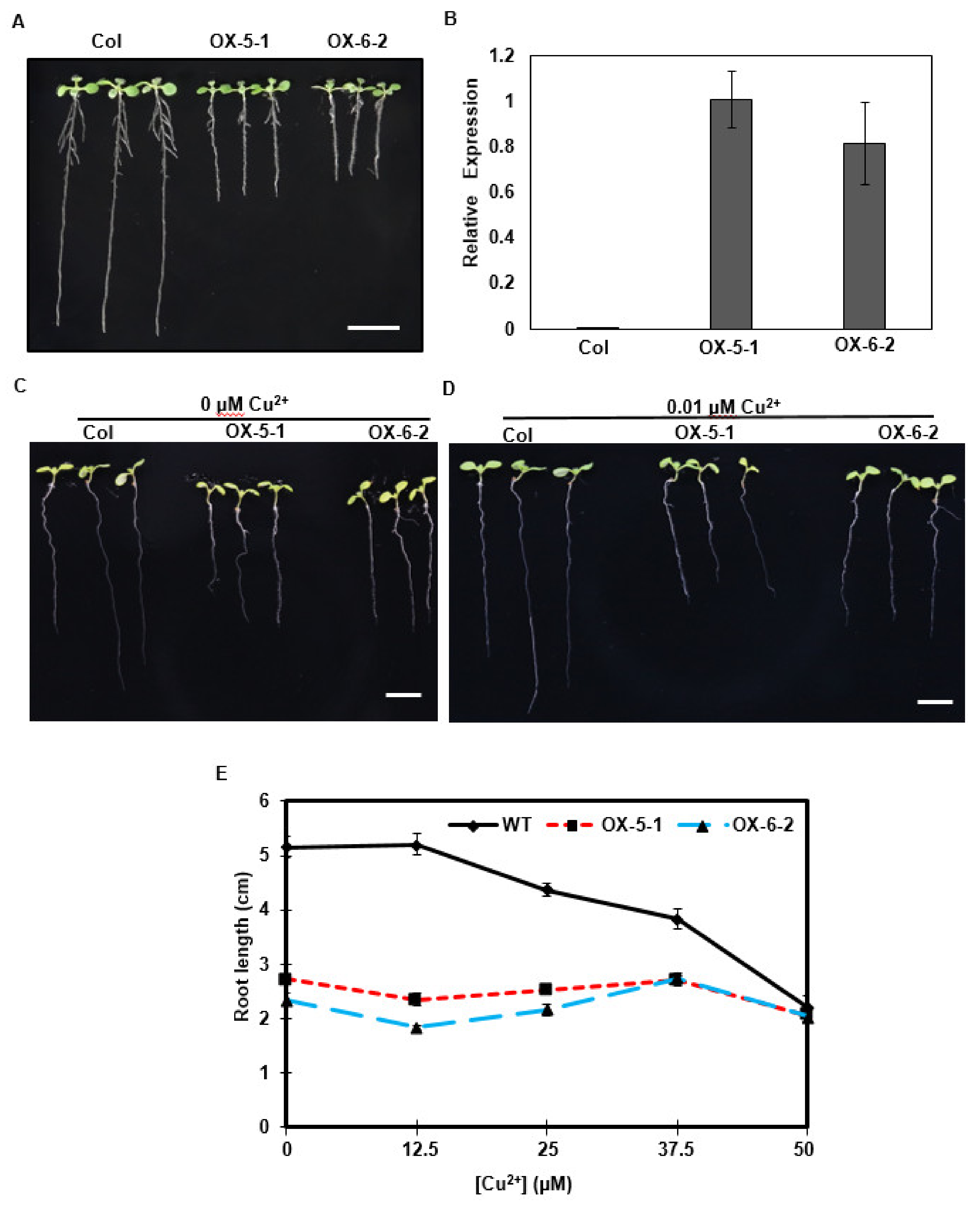
2.5. Overexpression of a Tea Gene Encoding P1B-Type ATPase 5 in Arabidopsis Resulted in Hyposensitivity to Copper
3. Materials and Methods
3.1. Plant Materials
3.2. Total RNA Extraction
3.3. mRNA Isolation
3.4. cDNA Synthesis
3.5. cDNA Denaturation, Hybridization, and DSN Treatment for Normalization
3.6. Amplification of Normalized cDNA
3.7. Sfi I Digestion and cDNA Size Fractionation
3.8. cDNA Ligation into λTriplEx2 Vector
3.9. Package Ligation Mixture
3.10. Titering the Unamplified Library and Determination of the Rate of Recombination
3.11. Library Amplification and Titering
3.12. Converting λTriplEx2 to pTriplEx2 Plasmid
3.13. cDNA Sequencing
3.14. Binary Vector pBIG2113SF-M Construction
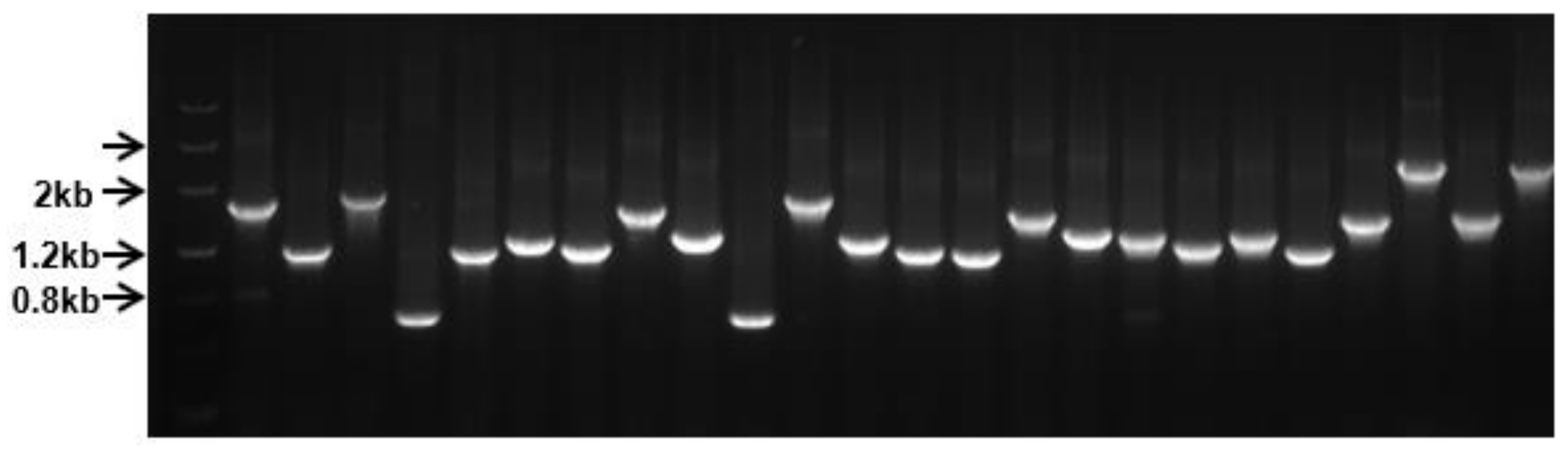
3.15. Cloning Tea FL-cDNAs into pBIG2113SF-M Vector
3.16. Transforming Tea FL-cDNA into Arabidopsis
3.17. Root Length Measurement and Gene Expression Analysis
3.18. Bioinformatic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Suzuki, Y.; Yoshitomo-Nakagawa, K.; Maruyama, K.; Suyama, A.; Sugano, S. Construction and characterization of a full length-enriched and a 5′-end-enriched cDNA library. Gene 1997, 200, 149–156. [Google Scholar] [CrossRef]
- Seki, M.; Narusaka, M.; Kamiya, A.; Ishida, J.; Satou, M.; Sakurai, T.; Nakajima, M.; Enju, A.; Akiyama, K.; Oono, Y.; et al. Functional annotation of a full-length Arabidopsis cDNA collection. Science 2002, 296, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.C.; Alonso-Cantabrana, H.; Forment, J.; Arribas, R.; Alamar, S.; Conejero, V.; Perez-Amador, M.A. A new set of ESTs and cDNA clones from full-length and normalized libraries for gene discovery and functional characterization in citrus. BMC Genom. 2009, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Makita, Y.; Shimada, S.; Kawashima, M.; Kondou-Kuriyama, T.; Toyoda, T.; Matsui, M. MOROKOSHI: Transcriptome database in sorghum bicolor. Plant Cell Physiol. 2015, 56, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Satoh, K.; Nagata, T.; Kawagashira, N.; Doi, K.; Kishimoto, N.; Yazaki, J.; Ishikawa, M.; Yamada, H.; Ooka, H.; et al. Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice. Science 2003, 301, 376–379. [Google Scholar] [CrossRef]
- Umezawa, T.; Sakurai, T.; Totoki, Y.; Toyoda, A.; Seki, M.; Ishiwata, A.; Akiyama, K.; Kurotani, A.; Yoshida, T.; Mochida, K.; et al. Sequencing and analysis of approximately 40,000 soybean cDNA clones from a full length-enriched cDNA library. DNA Res. 2008, 15, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Soderlund, C.; Descour, A.; Kudrna, D.; Bomhoff, M.; Boyd, L.; Currie, J.; Angelova, A.; Collura, K.; Wissotski, M.; Ashley, E.; et al. Sequencing, mapping, and analysis of 27,455 Maize full-length cDNAs. PLoS Genet. 2009, 5, e1000740. [Google Scholar] [CrossRef]
- Aoki, K.; Yano, K.; Suzuki, A.; Kawamura, S.; Sakurai, N.; Suda, K.; Kurabayashi, A.; Suzuki, T.; Tsugane, T.; Watanabe, M.; et al. Large-scale analysis of full-length cDNAs from the tomato (Solanum lycopersicum) cultivar Micro-Tom, a reference system for the Solanaceae genomics. BMC Genom. 2010, 11, 210. [Google Scholar] [CrossRef]
- Lin, M.; Lai, D.; Pang, C.; Fan, S.; Song, M.; Yu, S. Generation and analysis of a large-scale expressed sequence tag database from a full-length enriched cDNA library of developing leaves of Gossypium hirsutum L. PLoS ONE 2013, 8, e76443. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, H.; Qi, F.; Jian, G. Generation of transcriptome profiling and gene functional analysis in Gossypium hirsutum upon Verticillium dahliae infection. Biochem. Biophys. Res. Commun. 2016, 473, 879–885. [Google Scholar] [CrossRef]
- Ogihara, Y.; Mochida, K.; Kawaura, K.; Murai, K.; Seki, M.; Kamiya, A.; Shinozaki, K.; Carninci, P.; Hayashizaki, H.; Shin-I, T.; et al. Construction of a full-length cDNA library from young spikelets of hexaploid wheat and its characterization by large-scale sequencing of expressed sequence tags. Genes Genet. Syst. 2004, 79, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Shibata, Y.; Hayatsu, N.; Sugahara, Y.; Shibata, K.; Itoh, M.; Konno, H.; Okazaki, Y.; Muramatsu, M.; Hayashizaki, Y. Normalization and subtraction of CAP-trapper-selected cDNAs to prepare full-length cDNA libraries for rapid discovery of new genes. Genome Res. 2000, 10, 1617–1630. [Google Scholar] [CrossRef] [PubMed]
- Zhulidov, P.A.; Bogdanova, E.A.; Shcheglov, A.S.; Shagina, I.A.; Vagner, L.L.; Khazpekov, G.L.; Kozhemiako, V.V.; Luk’ianov, S.A.; Shagin, D.A. A method for the preparation of normalized cDNA libraries enriched with full-length sequences. Russ. J. Bioorg. Chem. 2005, 31, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Apoaroliswa, Z.; Chen, Z.M. Global Tea Breeding: Achievement, Challenges and Perspectives; Zhejiang University Press: Zhejiang, China, 2012. [Google Scholar]
- Di, T.; Zhao, L.; Chen, H.; Qian, W.; Wang, P.; Zhang, X.; Xia, T. Transcriptomic and metabolic insights into the distinctive effects of exogenous melatonin and gibberellin on terpenoid synthesis and plant hormone signal transduction pathway in Camellia sinensis. J. Agric. Food Chem. 2019, 67, 4689–4699. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhu, C.; Zhao, S.; Zhang, S.; Wang, W.; Fu, H.; Li, X.; Zhou, C.; Chen, L.; Lin, Y.; et al. De novo transcriptome and phytochemical analyses reveal differentially expressed genes and characteristic secondary metabolites in the original oolong tea (Camellia sinensis) cultivar ‘Tieguanyin’ compared with cultivar ‘Benshan’. BMC Genom. 2019, 20, 265. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, M.; Wu, X.; Li, D.; Borthakur, D.; Ye, J.; Zheng, X.; Lu, J. Analysis of differentially expressed genes in tissues of Camellia sinensis during dedifferentiation and root redifferentiation. Sci. Rep. 2019, 9, 2935. [Google Scholar] [CrossRef]
- Xin, Z.; Ge, L.; Chen, S.; Sun, X. Enhanced transcriptome responses in herbivore-infested tea plants by the green leaf volatile (Z)-3-hexenol. J. Plant Res. 2019, 132, 285–293. [Google Scholar] [CrossRef]
- Wu, L.; Fang, Z.; Lin, J.; Sun, Y.; Du, Z.; Guo, Y.; Liu, J.; Liang, Y.; Ye, J. Complementary iTRAQ proteomic and transcriptomic analyses of leaves in tea plant (Camellia sinensis L.) with different maturity and regulatory network of flavonoid biosynthesis. J. Proteome Res. 2019, 18, 252–264. [Google Scholar]
- Xia, E.; Zhang, H.; Sheng, J.; Li, K.; Zhang, Q.; Kim, C.; Zhang, Y.; Liu, Y.; Zhu, T.; Li, W.; et al. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol. Plant 2017, 10, 866–877. [Google Scholar] [CrossRef]
- Wei, C.; Yang, H.; Wang, S.; Zhao, J.; Liu, C.; Gao, L.; Xia, E.; Lu, Y.; Tai, Y.; She, G.; et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4145–E4158. [Google Scholar] [CrossRef]
- Qiao, D.; Yang, C.; Chen, J.; Guo, Y.; Li, Y.; Niu, S.; Cao, K.; Che, Z. Comprehensive identification of the full-length transcripts and alternative splicing related to the secondary metabolism pathways in the tea plant (Camellia sinensis). Sci. Rep. 2019, 9, 2709. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.K.; Bhattacharya, A.; Ahuja, P.S.; Chand, P.K. Factor effecting Agrobacterium tumefaciens mediated transformation of tea (Camellia sinensis (L). O. Kuntze. Plant Cell Rep. 2001, 20, 712–720. [Google Scholar] [CrossRef]
- Osato, N.; Yamada, H.; Satoh, K.; Ooka, H.; Yamamoto, M.; Suzuki, K.; Kawai, J.; Carninci, P.; Ohtomo, Y.; Murakami, K.; et al. Antisense transcripts with rice full-length cDNAs. Genome Biol. 2003, 5, R5. [Google Scholar] [CrossRef] [PubMed]
- Brandle, J.E.; Richman, A.; Swanson, A.K.; Chapman, B.P. Leaf ESTs from Stevia rebaudiana: A resource for gene discovery in diterpene synthesis. Plant Mol. Biol. 2002, 50, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Wang, X.W.; Xu, Q.S.; Zhao, S.Q.; Tai, Y.L.; Wei, C.L. Global dissection of alternative splicing uncovers transcriptional diversity in tissues and associates with the flavonoid pathway in tea plant (Camellia sinensis). BMC Plant Biol. 2018, 18, 266. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Lu, Y.; Zinta, G.; Lang, Z.; Zhu, J.K. UTR-Dependent Control of Gene Expressionin Plants. Trends Plant Sci. 2018, 23, 248–259. [Google Scholar] [CrossRef]
- Pang, P.P.; Meyerowitz, E.M. Arabidopsis thaliana: A model system for plant molecular biology. Nat. Biotechnol. 1987, 5, 1177–1181. [Google Scholar] [CrossRef]
- Taji, T.; Ohsumi, C.; Iuchi, S.; Seki, M.; Kasuga, M.; Kobayashi, M.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Important roles of drought- and cold-inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. Plant J. 2002, 29, 417–426. [Google Scholar] [CrossRef]
- Andrés-Colás, N.; Sancenón, V.; Rodríguez-Navarro, S.; Mayo, S.; Thiele, D.J.; Ecker, J.R.; Puig, S.; Peń arrubia, L. The Arabidopsis heavy metal P-type ATPase HMA5 interacts with metallochaperones and functions in copper detoxification of roots. Plant. J. 2006, 45, 225–236. [Google Scholar] [CrossRef]
- Lucila, G.; Elina, W.; Uta, G.; Agustín, L.A.; Iris, S.; Daniel, H.G. The cytochrome c oxidase biogenesis factor AtCOX17 modulates stress responses in Arabidopsis. Plant Cell Environ. 2016, 39, 628–644. [Google Scholar]
- Attallah, C.V.; Welchen, E.; Gonzalez, D.H. The promoters of Arabidopsis thaliana genes AtCOX17-1 and -2, encoding a copper chaperone involved in cytochrome c oxidase biogenesis, are preferentially active in roots and anthers and induced by biotic and abiotic stress. Physiol. Plant. 2007, 129, 123–134. [Google Scholar] [CrossRef]
- Klaumann, S.; Nickolaus, S.D.; Fürst, S.H.; Starck, S.; Schneider, S.; Ekkehard Neuhaus, H.; Trentmann, O. The tonoplast copper transporter COPT5 acts as an exporter and is required for interorgan allocation of copper in Arabidopsis thaliana. New Phytol. 2011, 192, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Binder, B.M.; Rodríguez, F.I.; Bleecker, A.B. The Copper Transporter RAN1 Is Essential for Biogenesis of Ethylene Receptors in Arabidopsis. J. Biol. Chem. 2010, 285, 37263–37270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Ghany, S.E.; Müller-Moulé, P.; Niyogi, K.K.; Pilon, M.; Shikanai, T. Two P-Type ATPases Are Required for Copper Delivery inArabidopsis thaliana Chloroplasts. Plant Cell 2005, 17, 1233–1251. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Iqbal, M.; Zhang, Q.; Spelt, C.; Bliek, M.; Hakvoort, H.W.J.; Quattrocchio, F.M.; Koes, R.; Schat, H. Two Silene vulgaris copper transporters residing in different cellular compartments confer copper hypertolerance by distinct mechanisms when expressed in Arabidopsis thaliana. New Phytol. 2017, 215, 1102–1114. [Google Scholar] [CrossRef] [Green Version]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Xia, E.; Li, F.; Tong, W.; Li, P.; Wu, Q.; Zhao, H.; Ge, R.; Li, R.; Li, Y.; Zhang, Z.; et al. Tea plant information archive (TPIA): A comprehensive genomics and bioinformatics platform for tea plant. Plant Biotechnol. J. 2019. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Carninci, P.; Kvam, C.; Kitamura, A.; Ohsumi, T.; Okazaki, Y.; Itoh, M.; Kamiya, M.; Shibata, K.; Sasaki, N.; Izawa, M.; et al. High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics 1996, 37, 327–336. [Google Scholar] [CrossRef]
- Edery, I.; Chu, L.; Sonenberg, N.; Pelletier, J. An efficient strategy to isolate full-length cDNAs based on an mRNA cap retention procedure (CAPture). Mol. Cell Biol. 1995, 15, 3363–3371. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.Y.; Machleder, E.M.; Chenchik, A.; Li, R.; Siebert, P.D. Reverse transcriptase template switching: A SMART (TM) approach for full-length cDNA library construction. Biotechniques 2001, 30, 892–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyavsky, A.; Vinogradova, T.V.; Rajewsky, K. PCR-based cDNA library construction: General cDNA libraries at the level of a few cells. Nucl. Acids Res. 1989, 17, 2919–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carninci, P.; Shibata, Y.; Hayatsu, N.; Itoh, M.; Shiraki, T.; Hirozane, T.; Watahiki, A.; Shibata, K.; Konno, H.; Muramatsu, M.; et al. Balanced-size and long-size cloning of full-length, cap-trapped cDNAs into vectors of the novel lambda-FLC family allows enhanced gene discovery rate and functional analysis. Genomics 2001, 77, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Nakazawa, M.; Kawashima, M.; Iizumi, H.; Kuroda, H.; Kondou, Y.; Tsuhara, Y.; Suzuki, K.; Ishikawa, A.; Seki, M.; et al. The FOX hunting system: An alternative gain-of-function gene hunting technique. Plant. J. 2006, 48, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Kondou, Y.; Higuchi, M.; Takahashi, S.; Sakurai, T.; Ichikawa, T.; Kuroda, H.; Yoshizumi, T.; Tsumoto, Y.; Horii, Y.; Kawashima, M.; et al. Systematic approaches to using the FOX hunting system to identify useful rice genes. Plant. J. 2009, 57, 883–894. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clone Number | Accession Number | CSA Number | CSS Number | 5′UTR (bp) | ORF (bp) | 3′UTR (bp) | Gene Annotation |
|---|---|---|---|---|---|---|---|
| 1 | MN027182 | CSA023243 | TEA018739 | 22 | 654 | 335 | polyadenylate-binding protein 2 isoform X1 |
| 2 | MK795745 | CSA033654 | TEA031121 | 33 | 912 | 505 | α-1,3/1,6-mannosyltransferase ALG2-like |
| 3 | MK795746 | CSA022245 | TEA026028 | 8 | 828 | 308 | protein COFACTOR ASSEMBLY OF COMPLEX C SUBUNIT B CCB2, isoform X7 |
| 4 | MN027183 | CSA027624 | TEA005502 | 73 | 1353 | 309 | vacuolar protein sorting-associated protein 9A-like |
| 5 | MK795747 | nd | TEA018613 | 194 | 1140 | 254 | F-box protein At3g07870-like |
| 6 | MK795748 | CSA033750 | TEA021240 | 81 | 459 | 459 | signal peptidase complex catalytic subunit SEC11A-like |
| 7 | MN027184 | CSA015215 | TEA028910 | 4 | 1158 | 213 | pentatricopeptide repeat-containing protein At5g50390, chloroplastic |
| 8 | MK795749 | CSA019466 | TEA000616 | 209 | 726 | 248 | Probable copper-transporting ATPase HMA5 |
| 9 | MN027185 | CSA017490 | TEA009722 | 93 | 315 | 280 | uncharacterized LOC114266360 (LOC114266360) |
| 10 | MK889351 | CSA017486 | TEA002539 | 102 | 768 | 276 | thioredoxin-like 2, chloroplastic |
| 11 | MK795750 | CSA028127 | TEA008577 | 157 | 579 | 258 | protein RER1B-like |
| 12 | MK795751 | CSA028739 | TEA024852 | 32 | 963 | 305 | probable serine/threonine-protein kinase At1g54610 |
| 13 | MN125540 | CSA004353 | TEA017437 | 45 | 1800 | 210 | LOW quality protein: BTB/POZ domain-containing protein At3g08570-like |
| 14 | MN102719 | CSA012903 | nd | 237 | 660 | 432 | LOW QUALITY PROTEIN: DExH-box ATP-dependent RNA helicase DExH9-like |
| 15 | MK795752 | CSA004217 | TEA003309 | 66 | 555 | 272 | protein EI24 homolog |
| 16 | MK889352 | CSA008914 | TEA006217 | 310 | 477 | 260 | LOW QUALITY PROTEIN: histone-lysine N-methyltransferase CLF |
| 17 | MN027187 | CSA015641 | TEA002353 | 61 | 555 | 181 | acid phosphatase 1 |
| 18 | MK795753 | CSA030107 | TEA029671 | 23 | 483 | 627 | ROOT primordium defective 1 |
| 19 | MK795754 | CSA023247 | TEA002601 | 206 | 1149 | 252 | E3 ubiquitin-protein ligase SIS3-like |
| 20 | MK795755 | nd | TEA024713 | 11 | 1059 | 194 | uncharacterized protein LOC114322803 isoform X2 |
| 21 | MK795756 | CSA012514 | TEA005305 | 19 | 954 | 200 | 2-oxoglutarate-dependent dioxygenase AOP3-like |
| 22 | MK795757 | nd | TEA001038 | 31 | 1374 | 351 | protein farnesyltransferase subunit beta isoform X2 |
| 23 | MK795758 | CSA018660 | TEA017439 | 28 | 1014 | 433 | protein XAP5 CIRCADIAN TIMEKEEPER |
| 24 | MK889353 | CSA006900 | TEA028663 | 383 | 591 | 259 | cinnamoyl-CoA reductase 1-like isoform X1 |
| 25 | MN027188 | CSA015703 | TEA028160 | 147 | 648 | 403 | uncharacterized protein LOC114291801 |
| 26 | MK795759 | CSA028933 | TEA016139 | 60 | 483 | 220 | 40S ribosomal protein S11 |
| 27 | MK795760 | CSA021019 | TEA026343 | 25 | 1077 | 142 | methylmalonate-semialdehyde dehydrogenase (acylating), mitochondrial-like isoform X3 |
| 28 | MK795761 | nd | TEA030658 | 88 | 516 | 332 | Universal stress protein A-like protein isoform X1 |
| 29 | MN027193 | CSA035528 | TEA027668 | 1013 | cylicin-1-like isoform X2 | ||
| 30 | MN027189 | CSA008021 | TEA020012 | 37 | 876 | 224 | B3 domain-containing transcription factor VRN1-like |
| 31 | MK795762 | nd | TEA005586 | 81 | 828 | 213 | uncharacterized LOC114272112, transcript variant X2, |
| 32 | MK795763 | CSA019133 | TEA015571 | 143 | 618 | 278 | uncharacterized protein LOC114308887 |
| 33 | MK795764 | nd | TEA019546 | 20 | 918 | 177 | uncharacterized LOC114312832 |
| 34 | MN158199 | nd | nd | Natural antisense RNA to CSA010175 or TEA005630 | |||
| 35 | MK795765 | CSA023599 | TEA024700 | 338 | 807 | 354 | uncharacterized LOC114281519, transcript variant X2 |
| 36 | MK795766 | CSA001233 | nd | 43 | 480 | 302 | Uncharacterized protein LOC104594327 |
| 37 | MK795767 | CSA031667 | TEA000191 | 363 | 666 | 262 | uncharacterized protein LOC114256570 |
| 38 | MN027190 | CSA000092 | TEA023002 | 11 | 645 | 341 | uncharacterized protein At4g15545-like |
| 39 | MN027191 | CSA017175 | TEA009315 | 152 | 879 | 540 | uncharacterized protein LOC114261191 isoform X2 |
| 40 | MN027192 | CSA029843 | TEA002496 | 601 | 750 | 305 | phospholipase A1-IIgamma-like (LOC114274378) |
| 41 | MK795768 | CSA009902 | TEA027481 | 66 | 651 | 256 | uncharacterized LOC114314960 |
| 42 | MK795769 | CSA026559 | TEA003389 | 243 | 768 | 39 | Uncharacterized protein |
| 43 | MN027194 | nd | nd | Noncoding RNA | |||
| 44 | MK889354 | nd | TEA023793 | 39 | 654 | 317 | RNA-binding protein 48-like isoform X2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, L.; Cai, W.; Du, Z.; Zhang, W.; Xu, Q.; Sun, W.; Chen, M. Establishing a System for Functional Characterization of Full-Length cDNAs of Camellia sinensis. Int. J. Mol. Sci. 2019, 20, 5929. https://doi.org/10.3390/ijms20235929
Lin L, Cai W, Du Z, Zhang W, Xu Q, Sun W, Chen M. Establishing a System for Functional Characterization of Full-Length cDNAs of Camellia sinensis. International Journal of Molecular Sciences. 2019; 20(23):5929. https://doi.org/10.3390/ijms20235929
Chicago/Turabian StyleLin, Lin, Weiwei Cai, Zhenghua Du, Wenjing Zhang, Quanming Xu, Weijiang Sun, and Mingjie Chen. 2019. "Establishing a System for Functional Characterization of Full-Length cDNAs of Camellia sinensis" International Journal of Molecular Sciences 20, no. 23: 5929. https://doi.org/10.3390/ijms20235929
APA StyleLin, L., Cai, W., Du, Z., Zhang, W., Xu, Q., Sun, W., & Chen, M. (2019). Establishing a System for Functional Characterization of Full-Length cDNAs of Camellia sinensis. International Journal of Molecular Sciences, 20(23), 5929. https://doi.org/10.3390/ijms20235929