Small Molecule Inhibitors Targeting the Heat Shock Protein System of Human Obligate Protozoan Parasites


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hsp90 Family
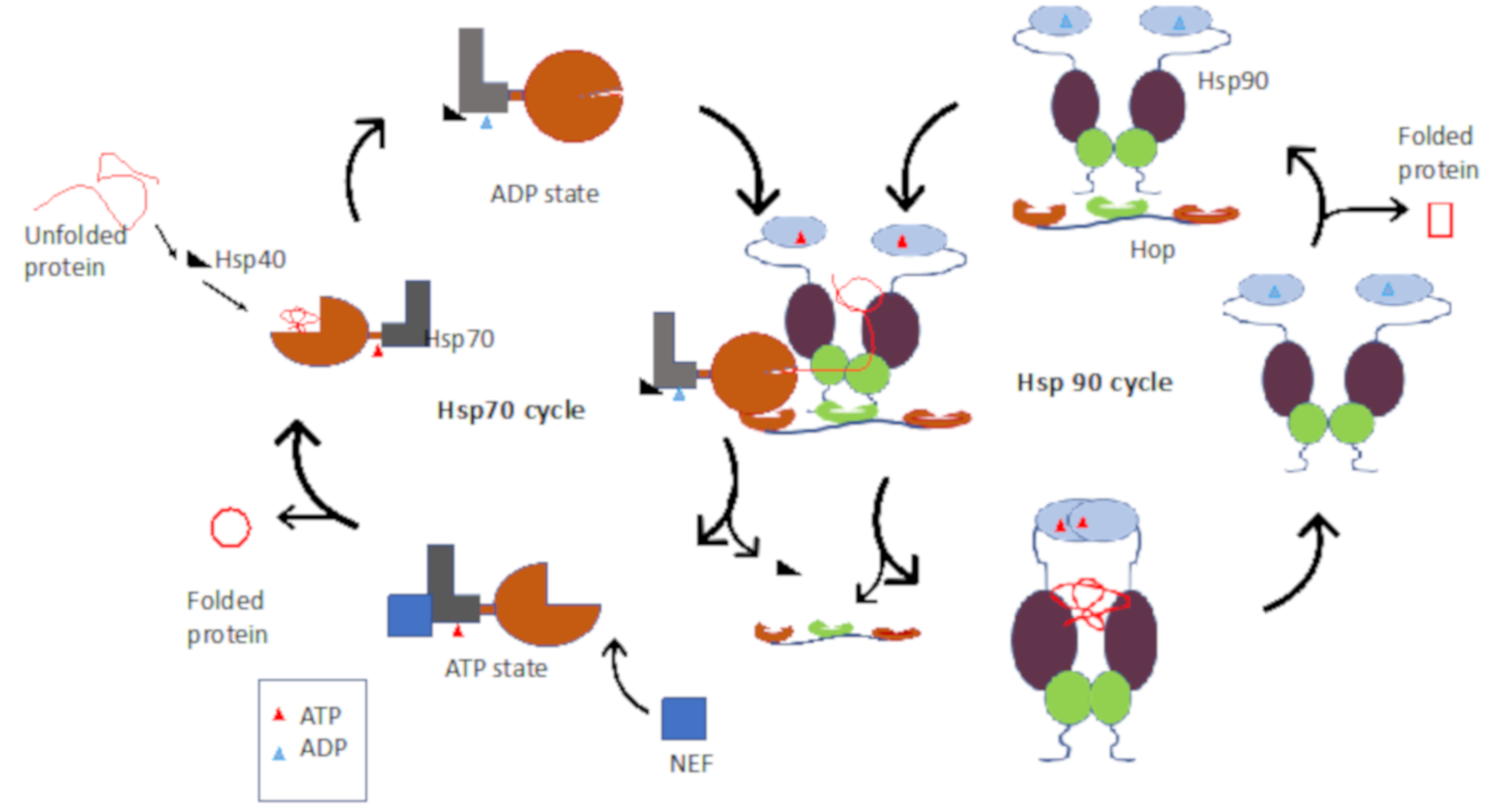
2.1. Hsp90–Hsp70 Chaperone Pathway
2.2. Hsp90 as a Drug Target
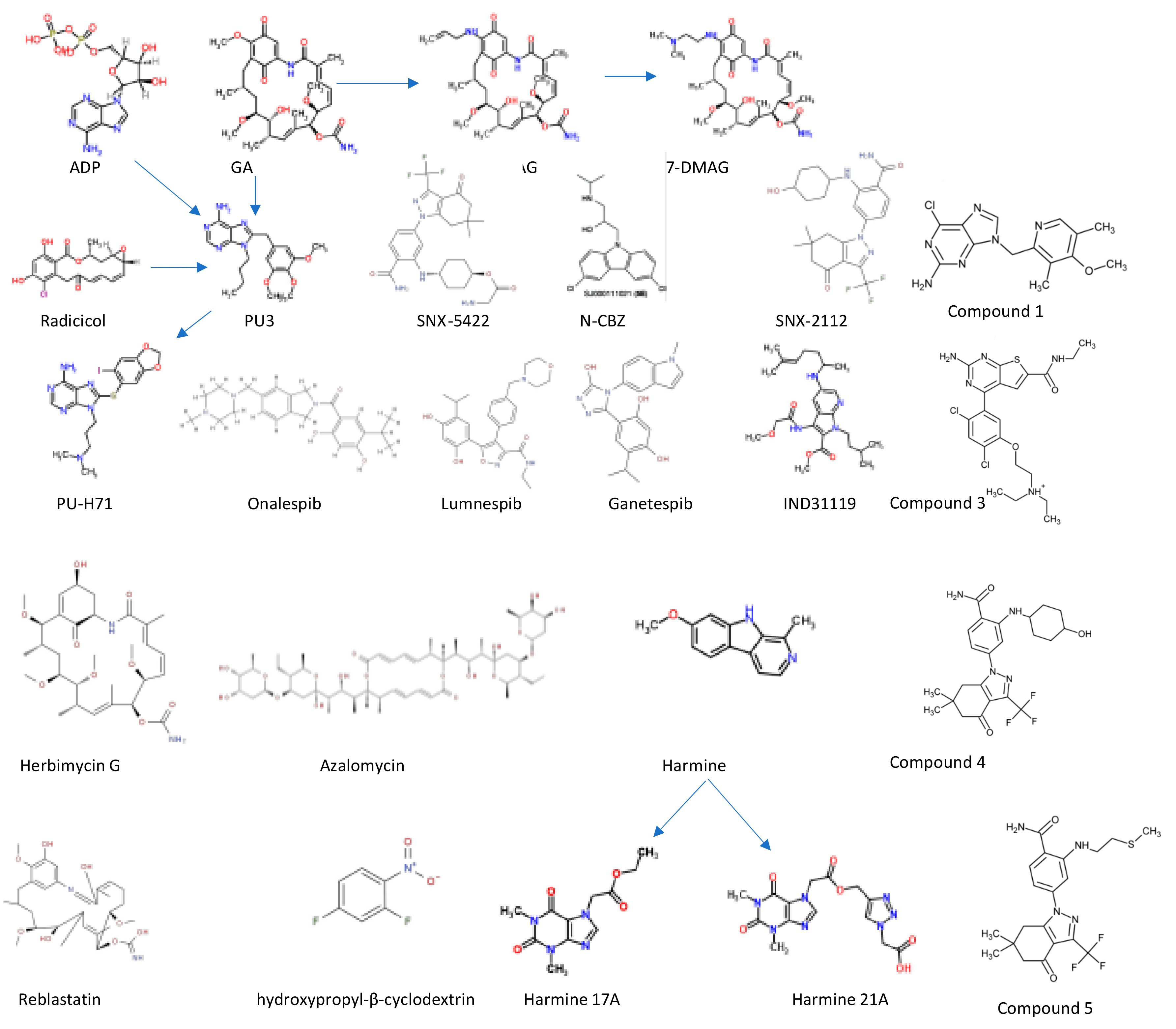
2.3. Small Molecule Inhibitors of Hsp90
2.4. Plasmodial Hsp90 as a Drug Target
2.5. Leishmanial Hsp90 as a Drug Target
2.6. Toxoplasma Hsp90 as a Drug Target
2.7. Trypanosoma Hsp90 as Drug Targets
3. Hsp100 Family
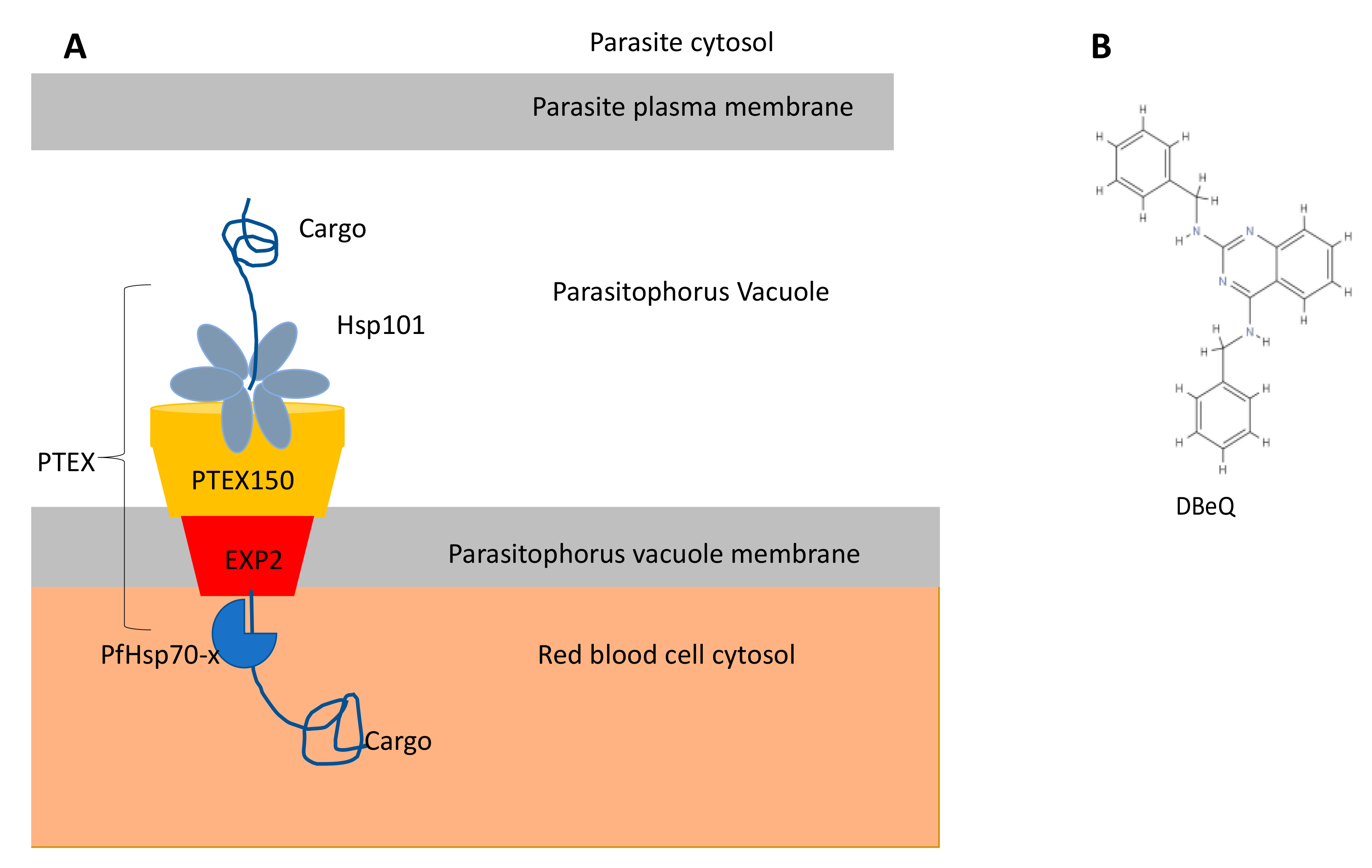
Parasitic Hsp100 as Drug Targets
4. Hsp60 Family
Targeting Parasite Hsp60
5. Hsp70 Chaperone System
P. falciparum Hsp70 as a Drug Target
6. Hsp40 Family of Molecular Chaperones
6.1. Hsp70–Hsp40 Partnership as a Drug Target
6.2. Targeting the P. falciparum Hsp70 and Hsp40 Partnership
7. Hsp110 Family of Proteins
7.1. Interaction of Hsp70 with Hsp110
7.2. Targeting the Hsp110–Hsp70 Interaction
8. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Shonhai, A.; Maier, A.G.; Przyborski, J.M.; Blatch, G.L. Intracellular protozoan parasites of humans: The role of molecular chaperones in development and pathogenesis. Protein Peptide Lett. 2011, 18, 143–157. [Google Scholar] [CrossRef]
- Mazumdar, J.; Wilson, E.H.; Masek, K.; Hunter, C.A.; Striepen, B. Apicoplast fatty acid synthesis is essential for organelle biogenesis and parasite survival in Toxoplasma gondii. Proc. Natl. Acad. Sci. USA 2006, 103, 13192–13197. [Google Scholar] [CrossRef]
- Foth, B.J.; Stimmler, L.M.; Handman, E.; Crabb, B.S.; Hodder, A.N.; McFadden, G.I. The malaria parasite Plasmodium falciparum has only one pyruvate dehydrogenase complex, which is located in the apicoplast. Mol. Microbiol. 2005, 55, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.; McFadden, G.I. The evolution, metabolism and functions of the apicoplast. Philos. Trans. R. Soc. B 2010, 365, 749–763. [Google Scholar] [CrossRef] [PubMed]
- McFadden, G.I. The apicoplast. Protoplasma 2011, 248, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.; van Dooren, G.G.; Agrawal, S.; Brooks, C.F.; McFadden, G.I.; Striepen, B.; Higgins, M.K. Tic22 is an essential chaperone required for protein import into the apicoplast. J. Biol. Chem. 2012, 287, 39505–39512. [Google Scholar] [CrossRef] [PubMed]
- Ochsenreiter, T.; Anderson, S.; Wood, Z.A.; Hajduk, S.L. Alternative RNA editing produces a novel protein involved in mitochondrial DNA maintenance in trypanosomes. Mol. Cell. Biol. 2008, 28, 5595–5604. [Google Scholar] [CrossRef]
- Agbe, A.; Yielding, K.L. Kinetoplasts play an important role in the drug responses of Trypanosoma brucei. J. Parasitol. 1995, 81, 968–973. [Google Scholar] [CrossRef]
- Caljon, G.; De Muylder, G.; Durnez, L.; Jennes, W.; Vanaerschot, M.; Dujardin, J.C. Alice in microbes’ land: Adaptations and counter-adaptations of vector-borne parasitic protozoa and their hosts. FEMS Microbiol. Rev. 2016, 40, 664–685. [Google Scholar] [CrossRef]
- Barrett, L.G.; Thrall, P.H.; Burdon, J.J.; Linde, C.C. Life history determines genetic structure and evolutionary potential of host–parasite interactions. Trends. Ecol. Evol. 2008, 23, 678–685. [Google Scholar] [CrossRef]
- Pallares, I.; Sanchez De Groot, N.; Iglesias, V.; Sant’Anna, R.; Biosca, A.; Fernandez-Busquets, X.; Ventura, S. Discovering putative prion-like proteins in Plasmodium falciparum: A computational and experimental analysis. Front. Microbiol. 2018, 9, 1737. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, M.D.; Martin, D.M.; Ferguson, M.A. Global quantitative SILAC phosphoproteomics reveals differential phosphorylation is widespread between the procyclic and bloodstream form lifecycle stages of Trypanosoma brucei. J. Proteome Res. 2013, 12, 2233–2244. [Google Scholar] [CrossRef] [PubMed]
- Butter, F.; Bucerius, F.; Michel, M.; Cicova, Z.; Mann, M.; Janzen, C.J. Comparative proteomics of two life cycle stages of stable isotope-labeled Trypanosoma brucei reveals novel components of the parasite’s host adaptation machinery. Mol. Cell. Proteomics 2013, 12, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.; Myler, P.J. Illuminating parasite protein production by ribosome profiling. Trends Parasitol. 2016, 32, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Folgueira, C.; Requena, J.M. A postgenomic view of the heat shock proteins in kinetoplastids. FEMS Microbiol. Rev. 2017, 31, 359–377. [Google Scholar] [CrossRef]
- Rao, A.; Kumar, M.K.; Joseph, T.; Bulusu, G. Cerebral malaria: Insights from host-parasite protein-protein interactions. Malar. J. 2010, 9, 155. [Google Scholar] [CrossRef]
- Cuesta Astroz, Y.; Santos, A.; Oliveira, G.C.D.; Jensen, L.J. Analysis of predicted host-parasite interactomes reveals commonalities and specificities related to parasitic lifestyle and tissues tropism. Front. Immunol. 2019, 10, 212. [Google Scholar] [CrossRef]
- Montagna, G.N.; Buscaglia, C.A.; Münter, S.; Goosmann, C.; Frischknecht, F.; Brinkmann, V.; Matuschewski, K. Critical role for heat shock protein 20 (HSP20) in migration of malarial sporozoites. J. Biol. Chem. 2012, 287, 2410–2422. [Google Scholar] [CrossRef]
- Biebl, M.M.; Buchner, J. Structure, function, and regulation of the Hsp90 machinery. CSH Perspect Biol. 2019, a034017. [Google Scholar] [CrossRef]
- Jackson, S.E. Hsp90: Structure and function. Top. Curr. Chem. 2013, 328, 155–240. [Google Scholar]
- Odunuga, O.O.; Hornby, J.A.; Bies, C.; Zimmermann, R.; Pugh, D.J.; Blatch, G.L. Tetratricopeptide repeat motif-mediated Hsc70-mSTI1 interaction molecular characterization of the critical contacts for successful binding and specificity. J. Biol. Chem. 2003, 278, 6896–6904. [Google Scholar] [CrossRef] [PubMed]
- Genest, O.; Wickner, S.; Doyle, S.M. Hsp90 and Hsp70 chaperones: Collaborators in protein remodeling. J. Biol. Chem. 2019, 294, 2109–2120. [Google Scholar] [CrossRef] [PubMed]
- Lässle, M.; Blatch, G.L.; Kundra, V.; Takatori, T.; Zetter, B.R. Stress-inducible, murine protein mSTI1 characterization of binding domains for heat shock proteins and in vitro phosphorylation by different kinases. J. Biol. Chem. 1997, 272, 1876–1884. [Google Scholar] [CrossRef]
- Röhl, A.; Wengler, D.; Madl, T.; Lagleder, S.; Tippel, F.; Herrmann, M.; Hendrix, J.; Richter, K.; Hack, G.; Schmid, A.B.; et al. Hsp90 regulates the dynamics of its cochaperone Sti1 and the transfer of Hsp70 between modules. Nat. Commun. 2015, 6, 6655. [Google Scholar]
- Johnson, B.D.; Schumacher, R.J.; Ross, E.D.; Toft, D.O. Hop modulates Hsp70/Hsp90 interactions in protein folding. J. Biol. Chem. 1998, 273, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.B.; Lagleder, S.; Gräwert, M.A.; Röhl, A.; Hagn, F.; Wandinger, S.K.; Cox, M.B.; Demmer, O.; Richter, K.; Groll, M.; et al. The architecture of functional modules in the Hsp90 co-chaperone Sti1/Hop. EMBO J. 2012, 31, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Smith, D.F. Hop as an adaptor in the heat shock protein 70 (Hsp70) and hsp90 chaperone machinery. J. Biol. Chem. 1998, 273, 35194–35200. [Google Scholar] [CrossRef]
- Zininga, T.; Makumire, S.; Gitau, G.W.; Njunge, J.M.; Pooe, O.J.; Klimek, H.; Scheurr, R.; Raifer, H.; Prinsloo, E.; Przyborski, J.M.; et al. Plasmodium falciparum Hop (PfHop) interacts with the Hsp70 chaperone in a nucleotide-dependent fashion and exhibits ligand selectivity. PLoS ONE 2015, 10, e0135326. [Google Scholar] [CrossRef]
- Kelley, W.L. Molecular chaperones: How J domains turn on Hsp70s. Curr. Biol. 1999, 9, R305–R308. [Google Scholar] [CrossRef]
- Craig, E.A.; Marszalek, J. How do J-proteins get Hsp70 to do so many different things? Trends Biochem. Sci. 2017, 42, 355–368. [Google Scholar] [CrossRef]
- Banumathy, G.; Singh, V.; Pavithra, S.R.; Tatu, U. Heat shock protein 90 function is essential for Plasmodium falciparum growth in human erythrocytes. J. Biol. Chem. 2003, 278, 18336–18345. [Google Scholar] [CrossRef] [PubMed]
- Wiesgigl, M.; Clos, J. Heat shock protein 90 homeostasis controls stage differentiation in Leishmania donovani. Mol. Biol. Cell 2001, 12, 3307–3316. [Google Scholar] [CrossRef] [PubMed]
- Angel, S.O.; Figueras, M.J.; Alomar, M.L.; Echeverria, P.C.; Deng, B. Toxoplasma gondii Hsp90: Potential roles in essential cellular processes of the parasite. Parasitology 2014, 141, 1138–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidera, K.; Patsavoudi, E. Hsp90 inhibitors: Current development and potential in cancer therapy. Recent Pat. Anti-Cancer Drug Discov. 2014, 9, 1–20. [Google Scholar] [CrossRef]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef]
- Vali, S.; Pallavi, R.; Kapoor, S.; Tatu, U. Virtual prototyping study shows increased ATPase activity of Hsp90 to be the key determinant of cancer phenotype. Syst. Synth. Biol. 2010, 4, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Pallavi, R.; Roy, N.; Nageshan, R.K.; Talukdar, P.; Pavithra, S.R.; Reddy, R.; Venketesh, S.; Kumar, R.; Gupta, A.K.; Singh, R.K.; et al. Heat shock protein 90 as a drug target against protozoan infections biochemical characterization of Hsp90 from Plasmodium falciparum and Trypanosoma evansi and evaluation of its inhibitor as a candidate drug. J. Biol. Chem. 2010, 285, 37964–37975. [Google Scholar] [CrossRef] [Green Version]
- Wider, D.; Péli-Gulli, M.P.; Briand, P.A.; Tatu, U.; Picard, D. The complementation of yeast with human or Plasmodium falciparum Hsp90 confers differential inhibitor sensitivities. Mol. Biochem. Parasitol. 2009, 164, 147–152. [Google Scholar] [CrossRef]
- Krukenberg, K.A.; Street, T.O.; Lavery, L.A.; Agard, D.A. Conformational dynamics of the molecular chaperone Hsp90. Q. Rev. Biophys. 2011, 44, 229–255. [Google Scholar] [CrossRef] [Green Version]
- Zuiderweg, E.R.; Hightower, L.E.; Gestwicki, J.E. The remarkable multivalency of the Hsp70 chaperones. Cell Stress Chaperones 2017, 22, 173–189. [Google Scholar] [CrossRef] [Green Version]
- Sahasrabudhe, P.; Rohrberg, J.; Biebl, M.M.; Rutz, D.A.; Buchner, J. The plasticity of the Hsp90 co-chaperone system. Mol. Cell. 2017, 67, 947–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.L.; Brown, C. Plasticity of the Hsp90 chaperone machine in divergent eukaryotic organisms. Cell Stress Chaperones 2009, 14, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botha, M.; Pesce, E.R.; Blatch, G.L. The Hsp40 proteins of Plasmodium falciparum and other apicomplexa: Regulating chaperone power in the parasite and the host. Int. J. Biochem. Cell Biol. 2007, 39, 1781–1803. [Google Scholar] [CrossRef] [PubMed]
- Njunge, J.H.; Ludewig, M.; Boshoff, A.; Pesce, E.R.; Blatch, G.L. Hsp70s and J proteins of Plasmodium parasites infecting rodents and primates: Structure, function, clinical relevance, and drug targets. Curr. Pharm. Des. 2013, 19, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Zhu, P.; Southall, N.; Inglese, J.; Austin, C.P.; Zheng, W.; Regan, L. An AlphaScreenTM-based high-throughput screen to identify inhibitors of Hsp90-cochaperone interaction. J. Biomol. Screen. 2009, 14, 273–281. [Google Scholar] [PubMed] [Green Version]
- Horibe, T.; Kohno, M.; Haramoto, M.; Ohara, K.; Kawakami, K. Designed hybrid TPR peptide targeting Hsp90 as a novel anticancer agent. J. Transl. Med. 2011, 9, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrent, M.; Pulido, D.; Rivas, L.; Andreu, D. Antimicrobial peptide action on parasites. Curr. Drug Targets 2012, 13, 1138–1147. [Google Scholar] [CrossRef]
- Marqus, S.; Pirogova, E.; Piva, T.J. Evaluation of the use of therapeutic peptides for cancer treatment. J. Biomed. Sci. 2017, 24, 21. [Google Scholar] [CrossRef] [Green Version]
- Dagan, A.; Efron, L.; Gaidukov, L.; Mor, A.; Ginsburg, H. In vitro antiplasmodium effects of dermaseptin S4 derivatives. Antimicrob. Agents Chemother. 2002, 46, 1059–1066. [Google Scholar] [CrossRef] [Green Version]
- Efron, L.; Dagan, A.; Gaidukov, L.; Ginsburg, H.; Mor, A. Direct interaction of dermaseptin S4 aminoheptanoyl derivative with intraerythrocytic malaria parasite leading to increased specific antiparasitic activity in culture. J. Biol. Chem. 2002, 277, 24067–24072. [Google Scholar] [CrossRef] [Green Version]
- Jhaveri, K.; Ochiana, S.O.; Dunphy, M.P.; Gerecitano, J.F.; Corben, A.D.; Peter, R.I.; Janjigian, Y.Y.; Gomes-DaGama, E.M.; Koren, J.; Modi, S.; et al. Heat shock protein 90 inhibitors in the treatment of cancer: Current status and future directions. Expert Opin. Investig. Drugs 2014, 23, 611–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515. [Google Scholar] [CrossRef] [PubMed]
- Shonhai, A. Plasmodial heat shock proteins: Targets for chemotherapy. FEMS Immunol. Med. Microbiol. 2010, 58, 61–74. [Google Scholar] [CrossRef]
- Pesce, E.R.; Cockburn, I.L.; Goble, J.L.; Stephens, L.L.; Blatch, G.L. Malaria heat shock proteins: Drug targets that chaperone other drug targets. Infect. Disord. Drug Targets 2010, 10, 147–157. [Google Scholar] [CrossRef]
- Posfai, D.; Eubanks, A.L.; Keim, A.I.; Lu, K.Y.; Wang, G.Z.; Hughes, P.F.; Kato, N.; Haystead, T.A.; Derbyshire, E.R. Identification of Hsp90 inhibitors with anti-Plasmodium activity. Antimicrob. Agents Chemother. 2018, 62, e01799-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murillo-Solano, C.; Dong, C.; Sanchez, C.G.; Pizarro, J.C. Identification and characterization of the antiplasmodial activity of Hsp90 inhibitors. Malar. J. 2017, 16, 292. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.J.; Caton, E.; Shapiro, T.A. Model system identifies kinetic driver of Hsp90 inhibitor activity against African trypanosomes and Plasmodium falciparum. Antimicrob. Agents Chemother. 2018, 62, e00056-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiosis, G. Targeting chaperones in transformed systems–a focus on Hsp90 and cancer. Expert Opin. Ther. Targets 2006, 10, 37–50. [Google Scholar] [CrossRef]
- Immormino, R.M.; Metzger, L.E., IV; Reardon, P.N.; Dollins, D.E.; Blagg, B.S.; Gewirth, D.T. Different poses for ligand and chaperone in inhibitor-bound Hsp90 and GRP94: Implications for paralog-specific drug design. J. Mol. Biol. 2009, 388, 1033–1042. [Google Scholar] [CrossRef] [Green Version]
- Shahinas, D.; Folefoc, A.; Pillai, D. Targeting Plasmodium falciparum Hsp90: Towards reversing antimalarial resistance. Pathogens 2013, 2, 33–54. [Google Scholar] [CrossRef] [Green Version]
- Horibe, T.; Torisawa, A.; Kohno, M.; Kawakami, K. Molecular mechanism of cytotoxicity induced by Hsp90-targeted Antp-TPR hybrid peptide in glioblastoma cells. Mol. Cancer 2012, 11, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuehlke, A.D.; Moses, M.A.; Neckers, L. Heat shock protein 90: Its inhibition and function. Philos. Trans. R. Soc. B 2017, 373, 20160527. [Google Scholar] [CrossRef] [Green Version]
- Goldman, J.W.; Raju, R.N.; Gordon, G.A.; El-Hariry, I.; Teofilivici, F.; Vukovic, V.M.; Bradley, R.; Karol, M.D.; Chen, Y.; Guo, W.; et al. A first in human, safety, pharmacokinetics, and clinical activity phase I study of once weekly administration of the Hsp90 inhibitor ganetespib (STA-9090) in patients with solid malignancies. BMC Cancer 2013, 13, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahinas, D.; MacMullin, G.; Benedict, C.; Crandall, I.; Pillai, D.R. Harmine is a potent antimalarial targeting Hsp90 and synergizes with chloroquine and artemisinin. Antimicrob. Agents Chemother. 2012, 56, 4207–4213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, P.; Kumar, R.; Tatu, U. Chaperoning a cellular upheaval in malaria: Heat shock proteins in Plasmodium falciparum. Mol. Biochem. Parasitol. 2007, 153, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Catz, S.D.; Johnson, J.L.; Babior, B.M. Characterization of the nucleotide-binding capacity and the ATPase activity of the PIP3-binding protein JFC1. Proc. Natl. Acad. Sci. USA 2001, 98, 11230–11235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Mäser, P.; Picard, D. Inhibition of Plasmodium falciparum Hsp90 contributes to the antimalarial activities of aminoalcohol-carbazoles. J. Med. Chem. 2016, 59, 6344–6352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 1997, 90, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Stebbins, C.E.; Russo, A.A.; Schneider, C.; Rosen, N.; Hartl, F.U.; Pavletich, N.P. Crystal structure of an Hsp90–geldanamycin complex: Targeting of a protein chaperone by an antitumor agent. Cell 1997, 89, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Schulte, T.W.; Akinaga, S.; Soga, S.; Sullivan, W.; Stensgard, B.; Toft, D.; Neckers, L.M. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones 1998, 3, 100. [Google Scholar] [CrossRef] [Green Version]
- Bartsch, K.; Hombach-Barrigah, A.; Clos, J. Hsp90 inhibitors radicicol and geldanamycin have opposing effects on Leishmania Aha1-dependent proliferation. Cell Stress Chaperones 2017, 22, 729–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisi, L.; McGuire, S.; Sharp, A.; Chiosis, G.; Navarra, P.; Feinstein, D.L.; Russo, C.D. The novel Hsp90 inhibitor, PU-H71, suppresses glial cell activation but weakly affects clinical signs of EAE. J. Neuroimmunol. 2013, 255, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Bisson, W.H.; Mäser, P.; Scapozza, L.; Picard, D. Differences in conformational dynamics between Plasmodium falciparum and human Hsp90 orthologues enable the structure-based discovery of pathogen-selective inhibitors. J. Med. Chem. 2014, 57, 2524–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buedenbender, L.; Robertson, L.; Lucantoni, L.; Avery, V.; Kurtböke, D.; Carroll, A. HSQC-TOCSY fingerprinting-directed discovery of antiplasmodial polyketides from the marine Ascidian-derived Streptomyces sp.(USC-16018). Mar. Drugs 2018, 16, 189. [Google Scholar] [CrossRef] [Green Version]
- Bayih, A.G.; Folefoc, A.; Mohon, A.N.; Eagon, S.; Anderson, M.; Pillai, D.R. In vitro and in vivo anti-malarial activity of novel harmine-analog heat shock protein 90 inhibitors: A possible partner for artemisinin. Malar. J. 2016, 15, 579. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhou, Y.; Yao, C.; Ma, X.; Wang, L.; Xu, W.; Wang, Z.; Qiao, Z. Apoptosis caused by Hsp90 inhibitor geldanamycin in Leishmania donovani during promastigote-to-amastigote transformation stage. Parasitol. Res. 2009, 105, 1539. [Google Scholar] [CrossRef]
- Hombach, A.; Ommen, G.; Sattler, V.; Clos, J. Leishmania donovani P23 protects parasites against Hsp90 inhibitor-mediated growth arrest. Cell Stress Chaperones 2015, 20, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Bifeld, E.; Lorenzen, S.; Bartsch, K.; Vasquez, J.J.; Siegel, T.N.; Clos, J. Ribosome profiling reveals Hsp90 inhibitor effects on stage-specific protein synthesis in Leishmania donovani. mSystems 2018, 3, e00214–e00218. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi-Ostad-Kalayeh, S.; Stahl, F.; Scheper, T.; Kock, K.; Herrmann, C.; Heleno Batista, F.A.; Borges, J.C.; Sasse, F.; Eichner, S.; Ongouta, J.; et al. Heat shock proteins revisited: Using a mutasynthetically generated reblastatin library to compare the inhibition of human and Leishmania HSP90s. Chem. Biol. Chem. 2018, 19, 562–574. [Google Scholar] [CrossRef]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. BBA Mol. Cell Res. 2012, 1823, 742–755. [Google Scholar] [CrossRef] [Green Version]
- Petersen, A.L.D.O.A.; Campos, T.A.; dos Santos Dantas, D.A.; de Souza Rebouças, J.; da Silva, J.C.; de Menezes, J.P.; Formiga, F.R.; de Melo, J.V.; Machado, G.; Veras, P.S. Encapsulation of the HSP-90 chaperone inhibitor 17-aag in stable liposome allow increasing the therapeutic index as assessed, in vitro, on Leishmania amazonensis amastigotes-hosted in mouse CBA macrophages. Front. Cell Infect. Microbiol. 2018, 8, 30. [Google Scholar] [CrossRef]
- Sun, H.; Zhuo, X.; Zhao, X.; Yang, Y.; Chen, X.; Yao, C.; Du, A. The heat shock protein 90 of Toxoplasma gondii is essential for invasion of host cells and tachyzoite growth. Parasite 2017, 24, 22. [Google Scholar] [CrossRef]
- Echeverria, P.C.; Matrajt, M.; Harb, O.S.; Zappia, M.P.; Costas, M.A.; Roos, D.S.; Dubremetz, J.F.; Angel, S.O. Toxoplasma gondii Hsp90 is a potential drug target whose expression and subcellular localization are developmentally regulated. J. Mol. Biol. 2005, 350, 723–734. [Google Scholar] [CrossRef]
- Meyer, K.J.; Shapiro, T.A. Potent antitrypanosomal activities of heat shock protein 90 inhibitors in vitro and in vivo. J. Infec. Dis. 2013, 208, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Graefe, S.E.; Wiesgigl, M.; Gaworski, I.; Macdonald, A.; Clos, J. Inhibition of Hsp90 in Trypanosoma cruzi induces a stress response but no stage differentiation. Eukaryot. Cell 2002, 1, 936–943. [Google Scholar] [CrossRef] [Green Version]
- Bao, R.; Lai, C.J.; Qu, H.; Wang, D.; Yin, L.; Zifcak, B.; Atoyan, R.; Wang, J.; Samson, M.; Forrester, J.; et al. CUDC-305, a novel synthetic HSP90 inhibitor with unique pharmacologic properties for cancer therapy. Clin. Cancer Res. 2009, 15, 4046–4057. [Google Scholar] [CrossRef]
- Pizarro, J.C.; Hills, T.; Senisterra, G.; Wernimont, A.K.; Mackenzie, C.; Norcross, N.R.; Ferguson, M.A.; Wyatt, P.G.; Gilbert, I.H.; Hui, R. Exploring the Trypanosoma brucei Hsp83 potential as a target for structure guided drug design. PLoS Negl. Trop. Dis. 2013, 7, e2492. [Google Scholar] [CrossRef] [Green Version]
- Parsell, D.A.; Kowal, A.S.; Singer, M.A.; Lindquist, S. Protein disaggregation mediated by heat-shock protein Hspl04. Nature 1994, 372, 475. [Google Scholar] [CrossRef]
- Mogk, A.; Kummer, E.; Bukau, B. Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation. Front. Mol. Biosci. 2015, 2, 22. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, Ł.; Leźnicki, P.; Morawiec, E.; Litwińczuk, N.; Liberek, K. Role of a conserved aspartic acid in nucleotide binding domain 1 (NBD1) of Hsp100 chaperones in their activities. Cell Stress Chaperones 2012, 17, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Shorter, J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 2011, 6, e26319. [Google Scholar] [CrossRef] [Green Version]
- Zolkiewski, M. A camel pass through the eye of a needle: Protein unfolding activity of Clp ATPases. Mol. Microbiol. 2006, 61, 1094–1100. [Google Scholar] [CrossRef] [Green Version]
- Winkler, J.; Tyedmers, J.; Bukau, B.; Mogk, A. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J. Cell Biol. 2012, 198, 387–404. [Google Scholar] [CrossRef] [Green Version]
- Mogk, A.; Tomoyasu, T.; Goloubinoff, P.; Rüdiger, S.; Röder, D.; Langen, H.; Bukau, B. Identification of thermolabile Escherichia coli proteins: Prevention and reversion of aggregation by DnaK and ClpB. EMBO J. 1999, 18, 6934–6949. [Google Scholar] [CrossRef]
- Johnston, D.M.; Miot, M.; Hoskins, J.R.; Wickner, S.; Doyle, S.M. Substrate discrimination by ClpB and Hsp104. Front. Mol. Biosci. 2017, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Doyle, S.M.; Wickner, S. Hsp104 and ClpB: Protein disaggregating machines. Trends Biochem. Sci. 2009, 34, 40–48. [Google Scholar] [CrossRef]
- El Bakkouri, M.; Pow, A.; Mulichak, A.; Cheung, K.L.; Artz, J.D.; Amani, M.; Fell, S.; de Koning-Ward, T.F.; Goodman, C.D.; McFadden, G.I.; et al. The Clp chaperones and proteases of the human malaria parasite Plasmodium falciparum. J. Mol. Biol. 2010, 404, 456–477. [Google Scholar] [CrossRef]
- Rathore, S.; Jain, S.; Sinha, D.; Gupta, M.; Asad, M.; Srivastava, A.; Narayanan, M.S.; Ramasamy, G.; Chauhan, V.S.; Gupta, D.; et al. Disruption of a mitochondrial protease machinery in Plasmodium falciparum is an intrinsic signal for parasite cell death. Cell Death Dis. 2011, 2, e231. [Google Scholar] [CrossRef]
- El Bakkouri, M.; Rathore, S.; Calmettes, C.; Wernimont, A.K.; Liu, K.; Sinha, D.; Asad, M.; Jung, P.; Hui, R.; Mohmmed, A.; et al. Structural insights into the inactive subunit of the apicoplast-localized caseinolytic protease complex of Plasmodium falciparum. J. Biol. Chem. 2013, 288, 1022–1031. [Google Scholar] [CrossRef] [Green Version]
- de Koning-Ward, T.F.; Gilson, P.R.; Boddey, J.A.; Rug, M.; Smith, B.J.; Papenfuss, A.T.; Sanders, P.R.; Lundie, R.J.; Maier, A.G.; Cowman, A.F.; et al. A newly discovered protein export machine in malaria parasites. Nature 2009, 459, 945–949. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Ma, C.; Oberli, A.; Zinz, A.; Engels, S.; Przyborski, J.M. Proteomic analysis of exported chaperone/co-chaperone complexes of P. falciparum reveals an array of complex protein-protein interactions. Sci. Rep. 2017, 7, 42188. [Google Scholar] [CrossRef]
- Ho, C.M.; Beck, J.R.; Lai, M.; Cui, Y.; Goldberg, D.E.; Egea, P.F.; Zhou, Z.H. Malaria parasite translocon structure and mechanism of effector export. Nature 2018, 561, 70. [Google Scholar] [CrossRef]
- AhYoung, A.P.; Koehl, A.; Cascio, D.; Egea, P.F. Structural mapping of the ClpB ATPases of Plasmodium falciparum: Targeting protein folding and secretion for antimalarial drug design. Protein Sci. 2015, 24, 1508–1520. [Google Scholar] [CrossRef] [Green Version]
- Matthews, K.M.; Kalanon, M.; de Koning-Ward, T.F. Uncoupling the threading and unfoldase actions of Plasmodium Hsp101 reveals differences in export between soluble and insoluble proteins. mBio 2019, 10, e01106–e01119. [Google Scholar] [CrossRef] [Green Version]
- Elsworth, B.; Matthews, K.; Nie, C.Q.; Kalanon, M.; Charnaud, S.C.; Sanders, P.R.; Chisholm, S.A.; Counihan, N.A.; Shaw, P.J.; Pino, P.; et al. PTEX is an essential nexus for protein export in malaria parasites. Nature 2014, 511, 587. [Google Scholar] [CrossRef]
- Chou, T.F.; Brown, S.J.; Minond, D.; Nordin, B.E.; Li, K.; Jones, A.C.; Chase, P.; Porubsky, P.R.; Stoltz, B.M.; Schoenen, F.J.; et al. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 4834–4839. [Google Scholar] [CrossRef] [Green Version]
- Gilson, P.R.; Chisholm, S.A.; Crabb, B.S.; de Koning-Ward, T.F. Host cell remodelling in malaria parasites: A new pool of potential drug targets. Int. J. Parasitol. 2017, 47, 119–127. [Google Scholar] [CrossRef]
- Cao, S.; Du, N.; Chen, H.; Pang, Y.; Zhang, Z.; Zheng, J.; Jia, H. Toxoplasma gondii Clp family protein: TgClpB1 plays a crucial role in thermotolerance. Oncotarget 2017, 8, 86117–86129. [Google Scholar] [CrossRef] [Green Version]
- Hübel, A.; Krobitsch, S.; Hörauf, A.; Clos, J. Leishmania major Hsp100 is required chiefly in the mammalian stage of the parasite. Mol. Cell. Biol. 1997, 17, 5987–5995. [Google Scholar] [CrossRef] [Green Version]
- Külzer, S.; Charnaud, S.; Dagan, T.; Riedel, J.; Mandal, P.; Pesce, E.R.; Blatch, G.L.; Crabb, B.S.; Gilson, P.R.; Przyborski, J.M. Plasmodium falciparum encoded exported Hsp70/hsp40 chaperone/co-chaperone complexes within the host erythrocyte. Cell. Microbiol. 2012, 14, 1784–1795. [Google Scholar] [CrossRef]
- Bullen, H.E.; Charnaud, S.C.; Kalanon, M.; Riglar, D.T.; Dekiwadia, C.; Kangwanrangsan, N.; Torii, M.; Tsuboi, T.; Baum, J.; Ralph, S.A.; et al. Biosynthesis, localization, and macromolecular arrangement of the Plasmodium falciparum translocon of exported proteins (PTEX). J. Biol. Chem. 2012, 287, 7871–7884. [Google Scholar] [CrossRef] [Green Version]
- Viitanen, P.V.; Lubben, T.H.; Reed, J.; Goloubinoff, P.; O’Keefe, D.P.; Lorimer, G.H. Chaperonin facilitated refolding of ribulosebisphosphate carboxylase and ATP hydrolysis by chaperonin 60 (groEL) are K+ dependent. Biochemistry 1990, 29, 5665–5671. [Google Scholar] [CrossRef]
- Cappello, F.; Conway de Macario, E.; Marasà, L.; Zummo, G.; Macario, A.J. Hsp60 expression, new locations, functions, and perspectives for cancer diagnosis and therapy. Cancer Biol. Ther. 2008, 7, 801–809. [Google Scholar] [CrossRef]
- McCallum, C.D.; Do, H.; Johnson, A.E.; Frydman, J. The interaction of the chaperonin tailless complex polypeptide 1 (TCP1) ring complex (TRiC) with ribosome-bound nascent chains examined using photo-cross-linking. J. Cell Biol. 2000, 149, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar Mitra, K.; Kuldeep, J.; Siddiqi, M.I.; Goyal, N. The TCP1- γ subunit of Leishmania donovani forms a biologically active homo-oligomeric complex. FEBS J. 2015, 282, 4607–4619. [Google Scholar] [CrossRef] [Green Version]
- Ellis, R.J. Chaperonins: Introductory perspective. In The Chaperonins; Academic Press: Cambridge, MA, USA, 1996; pp. 1–25. [Google Scholar]
- Ellis, R.J. Chaperomics: In vivo GroEL function defined. Curr. Biol. 2005, 15, R661–R663. [Google Scholar] [CrossRef] [Green Version]
- Houry, W.A.; Frishman, D.; Eckerskorn, C.; Lottspeich, F.; Hartl, F.U. Identification of in vivo substrates of the chaperonin GroEL. Nature 1999, 402, 147. [Google Scholar] [CrossRef]
- Inobe, T.; Takahashi, K.; Maki, K.; Enoki, S.; Kamagata, K.; Kadooka, A.; Arai, M.; Kuwajima, K. Asymmetry of the GroEL-GroES complex under physiological conditions as revealed by small-angle X-ray scattering. Biophys. J. 2008, 94, 1392–1402. [Google Scholar] [CrossRef] [Green Version]
- Clare, D.K.; Vasishtan, D.; Stagg, S.; Quispe, J.; Farr, G.W.; Topf, M.; Horwich, A.L.; Saibil, H.R. ATP-triggered conformational changes delineate substrate-binding and-folding mechanics of the GroEL chaperonin. Cell 2012, 149, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Vilasi, S.; Bulone, D.; Caruso Bavisotto, C.; Campanella, C.; Marino Gammazza, A.; San Biagio, P.L.; Capello, F.; Conway de Marcario, E.; Macario, A.J. Chaperonin of group I: Oligomeric spectrum and biochemical and biological implications. Front. Mol. Biosci. 2018, 4, 99. [Google Scholar] [CrossRef]
- Lin, Z.; Schwarz, F.P.; Eisenstein, E. The hydrophobic nature of GroEL-substrate binding. J. Biol. Chem. 1995, 270, 1011–1014. [Google Scholar] [CrossRef] [Green Version]
- Ueno, T.; Taguchi, H.; Tadakuma, H.; Yoshida, M.; Funatsu, T. GroEL mediates protein folding with a two successive timer mechanism. Mol. Cell 2004, 14, 423–434. [Google Scholar] [CrossRef]
- Stevens, M.; Abdeen, S.; Salim, N.; Ray, A.M.; Washburn, A.; Chitre, S.; Sivinski, J.; Park, Y.; Hoang, Q.Q.; Chapman, E.; et al. Hsp60/10 chaperonin systems are inhibited by a variety of approved drugs, natural products, and known bioactive molecules. Bioorg. Med. Chem. Lett. 2019, 29, 1106–1112. [Google Scholar] [CrossRef]
- Abdeen, S.; Salim, N.; Mammadova, N.; Summers, C.M.; Goldsmith-Pestana, K.; McMahon-Pratt, D.; Schultz, P.G.; Horwich, A.L.; Chapman, E.; Johnson, S.M. Targeting the Hsp60/10 chaperonin systems of Trypanosoma brucei as a strategy for treating African sleeping sickness. Bioorg. Med. Chem. Lett. 2016, 26, 5247–5253. [Google Scholar] [CrossRef] [Green Version]
- Barrett, M.P.; Boykin, D.W.; Brun, R.; Tidwell, R.R. Human African trypanosomiasis: Pharmacological re-engagement with a neglected disease. Br. J. Pharmacol. 2007, 152, 1155–1171. [Google Scholar] [CrossRef]
- Sullivan, M.A.; Olson, C.L.; Winquist, A.G.; Engman, D.M. Expression and localization of Trypanosoma cruzi Hsp60. Mol. Biochem. Parasitol. 1994, 68, 197–208. [Google Scholar] [CrossRef]
- Droll, D.; Minia, I.; Fadda, A.; Singh, A.; Stewart, M.; Queiroz, R.; Clayton, C. Post-transcriptional regulation of the trypanosome heat shock response by a zinc finger protein. PLoS Pathog. 2013, 9, e1003286. [Google Scholar] [CrossRef] [Green Version]
- Willson, M.; Callens, M.; Kuntz, D.A.; Perié, J.; Opperdoes, F.R. Synthesis and activity of inhibitors highly specific for the glycolytic enzymes from Trypanosoma brucei. Mol. Biochem. Parasitol. 1993, 59, 201–210. [Google Scholar] [CrossRef]
- Stromberg, B.E.; Schlotthauer, J.C.; Conboy, G.A. The efficacy of closantel against Fascioloides magna in sheep. J. Parasitol. 1984, 70, 446. [Google Scholar] [CrossRef]
- Johnson, S.M.; Sharif, O.; Mak, P.A.; Wang, H.T.; Engels, I.H.; Brinker, A.; Schultz, P.G.; Horwich, A.L.; Chapman, E. A biochemical screen for GroEL/GroES inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 786–789. [Google Scholar] [CrossRef]
- Meng, Q.; Li, B.X.; Xiao, X. Toward developing chemical modulators of Hsp60 as potential therapeutics. Front. Mol. Biosci. 2018, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Nagumo, Y.; Kakeya, H.; Shoji, M.; Hayashi, Y.; Dohmae, N.; Osada, H. Epolactaene binds human Hsp60 Cys442 resulting in the inhibition of chaperone activity. Biochem. J. 2005, 387, 835–840. [Google Scholar] [CrossRef] [Green Version]
- Polson, E.S.; Kuchler, V.B.; Abbosh, C.; Ross, E.M.; Mathew, R.K.; Beard, H.A.; Williams, J.; Griffiths, H.B.S.; Shao, H.; Patel, A.; et al. KHS101 disrupts energy metabolism in human glioblastoma cells and reduces tumor growth in mice. Sci. Transl. Med. 2018, 10, eaar2718. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Wilson, R.J.M. The plastid of Plasmodium spp.: A target for inhibitors. In Malaria: Drugs, Disease and Post-Genomic Biology; Springer: Berlin/Heidelberg, Germany, 2005; pp. 251–273. [Google Scholar]
- Yeo, S.J.; Liu, D.X.; Park, H. Potential interaction of Plasmodium falciparum Hsp60 and Calpain. Korean J. Parasitol. 2015, 53, 665. [Google Scholar] [CrossRef]
- Tanabe, M.; Ishida, R.; Izuhara, F.; Komatsuda, A.; Wakui, H.; Sawada, K.; Otaka, M.; Nakamura, N.; Itoh, H. The ATPase activity of molecular chaperone Hsp60 is inhibited by immunosuppressant mizoribine. Am. J. Mol. Biol. 2012, 2, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Toursel, C.; Dzierszinski, F.; Bernigaud, A.; Mortuaire, M.; Tomavo, S. Molecular cloning, organellar targeting and developmental expression of mitochondrial chaperone Hsp60 in Toxoplasma gondii. Mol. Biochem. Parasitol. 2000, 111, 319–332. [Google Scholar] [CrossRef]
- Ashwinder, K.; Kho, M.T.; Chee, P.M.; Lim, W.Z.; Yap, I.K.; Choi, S.B.; Yam, W.K. Targeting heat shock proteins 60 and 70 of toxoplasma gondii as a potential drug target: In silico approach. Interdiscip. Sci. 2016, 8, 374–387. [Google Scholar] [CrossRef]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef]
- Easton, D.P.; Kaneko, Y.; Subjeck, J.R. The Hsp110 and Grp170 stress proteins: Newly recognized relatives of the Hsp70s. Cell Stress Chaperones 2000, 5, 276–290. [Google Scholar] [CrossRef] [Green Version]
- Sharma, Y.D. Structure and possible function of heat-shock proteins in Plasmodium falciparum. Comp. Biochem. Physiol. B 1992, 102, 437–444. [Google Scholar] [CrossRef]
- Chakafana, G.; Zininga, T.; Shonhai, A. Comparative structure-function features of Hsp70s of Plasmodium falciparum and human origins. Biophys. Rev. 2019, 11, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Sourjik, V. Physical map and dynamics of the chaperone network in Escherichia coli. Mol. Microbiol. 2012, 84, 736–747. [Google Scholar] [CrossRef]
- Alderson, T.R.; Kim, J.H.; Cai, K.; Frederick, R.O.; Tonelli, M.; Markley, J.L. The specialized Hsp70 (HscA) interdomain linker binds to its nucleotide-binding domain and stimulates ATP hydrolysis in both cis and trans configurations. Biochemistry 2014, 53, 7148–7159. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.P.; Schröder, H.; Rüdiger, S.; Paal, K.; Laufen, T.; Bukau, B. Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat. Struct. Biol. 2000, 7, 586–593. [Google Scholar]
- Kityk, R.; Kopp, J.; Sinning, I.; Mayer, M.P. Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol. Cell 2012, 48, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Needham, P.G.; Patel, H.J.; Chiosis, G.; Thibodeau, P.H.; Brodsky, J.L. Mutations in the Yeast Hsp70, Ssa1, at P417 alter ATP cycling, interdomain coupling, and specific chaperone functions. J. Mol. Biol. 2015, 427, 2948–2965. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.Y.; Lee, S.; Cyr, D.M. Mechanisms for regulation of Hsp70 function by Hsp40. Cell Stress Chaperones 2003, 8, 309. [Google Scholar] [CrossRef]
- Summers, D.W.; Douglas, P.M.; Ramos, C.H.; Cyr, D.M. Polypeptide transfer from Hsp40 to Hsp70 molecular chaperones. Trends Biochem. Sci. 2009, 34, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Kampinga, H.H.; Craig, E.A. The Hsp70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [Green Version]
- Cockburn, I.L.; Boshoff, A.; Pesce, E.R.; Blatch, G.L. Selective modulation of plasmodial Hsp70s by small molecules with antimalarial activity. Biol. Chem. 2014, 395, 1353–1362. [Google Scholar] [CrossRef]
- Mayer, M.P. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci. 2013, 38, 507–514. [Google Scholar] [CrossRef]
- Shonhai, A.; Boshoff, A.; Blatch, G.L. The structural and functional diversity of Hsp70 proteins from Plasmodium falciparum. Protein Sci. 2007, 16, 1803–1818. [Google Scholar] [CrossRef] [Green Version]
- Daniyan, M.O.; Przyborski, J.M.; Shonhai, A. Partners in mischief: Functional networks of heat shock proteins of Plasmodium falciparum and their influence on parasite virulence. Biomolecules 2019, 9, 295. [Google Scholar] [CrossRef] [Green Version]
- Dragovic, Z.; Broadley, S.A.; Shomura, Y.; Bracher, A.; Hartl, F.U. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J. 2006, 25, 2519–2528. [Google Scholar] [CrossRef] [Green Version]
- Zininga, T.; Achilonu, I.; Hoppe, H.; Prinsloo, E.; Dirr, H.W.; Shonhai, A. Overexpression, purification and characterisation of the Plasmodium falciparum Hsp70-z (PfHsp70-z) protein. PLoS ONE 2015, 10, e0129445. [Google Scholar] [CrossRef] [Green Version]
- Zininga, T.; Achilonu, I.; Hoppe, H.; Prinsloo, E.; Dirr, H.W.; Shonhai, A. Plasmodium falciparum Hsp70-z, an Hsp110 homologue, exhibits independent chaperone activity and interacts with Hsp70-1 in a nucleotide-dependent fashion. Cell Stress Chaperones 2016, 21, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Shonhai, A.; Botha, M.; de Beer, T.A.; Boshoff, A.; Blatch, G.L. Structure-function study of a Plasmodium falciparum Hsp70 using three-dimensional modelling and in vitro analyses. Protein Pept. Lett. 2008, 15, 1117–1125. [Google Scholar] [CrossRef]
- Shonhai, A.; Boshoff, A.; Blatch, G.L. Plasmodium falciparum heat shock protein 70 is able to suppress the thermosensitivity of an Escherichia coli DnaK mutant strain. Mol. Genet. Genom. 2005, 274, 70–78. [Google Scholar] [CrossRef]
- Kappes, B.; Suetterlin, B.W.; Hofer-Warbinek, R.; Humar, R.; Franklin, R.M. Two major phosphoproteins of Plasmodium falciparum are heat shock proteins. Mol. Biochem. Parasitol. 1993, 59, 83–94. [Google Scholar] [CrossRef]
- Bell, S.L.; Chiang, A.N.; Brodsky, J.L. Expression of a malarial Hsp70 improves defects in chaperone-dependent activities in ssa1 mutant yeast. PLoS ONE 2011, 6, e20047. [Google Scholar] [CrossRef] [Green Version]
- Stephens, L.L.; Shonhai, A.; Blatch, G.L. Co-expression of the Plasmodium falciparum molecular chaperone, PfHsp70, improves the heterologous production of the antimalarial drug target GTP cyclohydrolase I., PfGCHI. Prot. Expr. Purif. 2011, 77, 159–165. [Google Scholar] [CrossRef]
- Cockburn, I.L.; Pesce, E.-R.; Pryzborski, J.M.; Davies-Coleman, M.T.; Clark, P.G.; Keyzers, R.A.; Stephens, L.L.; Blatch, G.L. Screening for small molecule modulators of Hsp70 chaperone activity using protein aggregation suppression assays: Inhibition of the plasmodial chaperone PfHsp70-1. Biol. Chem. 2011, 392, 431–438. [Google Scholar] [CrossRef]
- Kapadia, G.J.; Azuine, M.A.; Balasubramanian, V.; Sridhar, R. Aminonaphthoquinones—A novel class of compounds with potent antimalarial activity against Plasmodium falciparum. Pharmacol. Res. 2001, 43, 363–367. [Google Scholar] [CrossRef]
- Zininga, T.; Pooe, O.J.; Makhado, P.B.; Ramatsui, L.; Prinsloo, E.; Achilonu, I.; Dirr, H.; Shonhai, A. Polymyxin B inhibits the chaperone activity of Plasmodium falciparum Hsp70. Cell Stress Chaperones 2017, 22, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Zininga, T.; Ramatsui, L.; Makhado, P.; Makumire, S.; Achilinou, I.; Hoppe, H.; Dirr, H.; Shonhai, A. (−)-Epigallocatechin-3-gallate inhibits the chaperone activity of Plasmodium falciparum Hsp70 chaperones and abrogates their association with functional partners. Molecules 2017, 22, 2139. [Google Scholar] [CrossRef] [Green Version]
- Sommer, M.S.; Gould, S.B.; Lehmann, P.; Gruber, A.; Przyborski, J.M.; Maier, U.G. Der1- mediated preprotein import into the periplastid compartment of chromalveolates? Mol. Biol. Evol. 2007, 24, 918–928. [Google Scholar] [CrossRef] [Green Version]
- Shonhai, A. Role of Hsp70s in development and pathogenicity of plasmodium species. In Heat Shock Proteins of Malaria; Shonhai, A., Blatch, G., Eds.; Springer: New York, NY, USA, 2014; pp. 47–70. [Google Scholar]
- Chen, Y.; Murillo-Solano, C.; Kirkpatrick, M.; Antoshchenko, T.; Park, T.; Pizarro, J. Repurposing drugs to target the malaria parasite unfolding protein response. Sci. Rep. 2018, 8, 10333–10350. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Li, X.; Lee, H.F.; Jinwal, U.K.; Srinivasan, S.R.; Seguin, S.P.; Young, Z.T.; Brodsky, J.L.; Dickey, C.A.; Sun, D.; et al. Synthesis and initial evaluation of YM-08, a blood-brain barrier permeable derivative of the heat shock protein 70 (Hsp70) inhibitor MKT-077, which reduces tau levels. ACS Chem. Neurosci. 2013, 4, 930–939. [Google Scholar] [CrossRef] [Green Version]
- Wickham, M.E.; Rug, M.; Ralph, S.A.; Klonis, N.; McFadden, G.I.; Tilley, L.; Cowman, A.F. Trafficking and assembly of the cytoadherence complex in Plasmodium falciparum-infected human erythrocytes. EMBO J. 2001, 20, 5636–5649. [Google Scholar] [CrossRef] [Green Version]
- Behl, A.; Kumar, V.; Bisht, A.; Panda, J.J.; Hora, R.; Mishra, P.C. Cholesterol bound Plasmodium falciparum co-chaperone ‘PFA0660w’complexes with major virulence factor ‘PfEMP1’via chaperone ‘PfHsp70-x’. Sci. Rep. 2019, 9, 2664. [Google Scholar] [CrossRef]
- Cobb, D.W.; Florentin, A.; Fierro, M.A.; Krakowiak, M.; Moore, J.M.; Muralidharan, V. The exported chaperone PfHsp70x is dispensable for the Plasmodium falciparum intraerythrocytic life cycle. mSphere 2017, 2, e00363-17. [Google Scholar] [CrossRef] [Green Version]
- Charnaud, S.C.; Dixon, M.W.; Nie, C.Q.; Chappell, L.; Sanders, P.R.; Nebl, T.; Hanssen, E.; Berriman, M.; Chan, J.A.; Blanch, A.J.; et al. The exported chaperone Hsp70-x supports virulence functions for Plasmodium falciparum blood stage parasites. PLoS ONE 2017, 12, e0181656. [Google Scholar] [CrossRef] [Green Version]
- Mabate, B.; Zininga, T.; Ramatsui, L.; Makumire, S.; Achilonu, I.; Dirr, H.W.; Shonhai, A. Structural and biochemical characterization of Plasmodium falciparum Hsp70-x reveals functional versatility of its C-terminal EEVN motif. Proteins 2018, 86, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Laufen, T.; Mayer, M.P.; Beisel, C.; Klostermeier, D.; Reinstein, J.; Bukau, B. Mechanism of regulation of Hsp70 chaperones by DnaJ co-chaperones. Proc. Natl. Acad. Sci. USA 1999, 96, 5452–5457. [Google Scholar] [CrossRef] [Green Version]
- Botha, M.; Chiang, A.N.; Needham, P.G.; Stephens, L.L.; Hoppe, H.C.; Külzer, S.; Przyborski, J.M.; Lingelbach, K.; Wipf, P.; Brodsky, J.L.; et al. Plasmodium falciparum encodes a single cytosolic type I Hsp40 that functionally interacts with Hsp70 and is upregulated by heat shock. Cell Stress Chaperones 2011, 16, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Rüdiger, S.; Schneider-Mergener, J.; Bukau, B. Its substrate specificity characterizes the DnaJ co-chaperone as a scanning factor for the DnaK chaperone. EMBO J. 2001, 20, 1042–1050. [Google Scholar] [CrossRef] [Green Version]
- Cheetham, M.E.; Caplan, A.J. Structure, function and evolution of DnaJ: Conservation and adaptation of chaperone function. Cell Stress Chaperones 1998, 3, 28–36. [Google Scholar] [CrossRef]
- Schilke, B.A.; Ciesielski, S.J.; Ziegelhoffer, T.; Kamiya, E.; Tonelli, M.; Lee, W.; Cornilescu, G.; Hines, J.K.; Markley, J.L.; Craig, E.A. Broadening the functionality of a J-protein/Hsp70 molecular chaperone system. PLoS Genet. 2017, 13, e1007084. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Maier, A.G.; Rug, M.; O’Neill, M.T.; Brown, M.; Chakravorty, S.; Szestak, T.; Chesson, J.; Wu, Y.; Hughes, K.; Coppel, R.L.; et al. Exported proteins required for virulence and rigidity of Plasmodium falciparum infected human erythrocytes. Cell 2008, 134, 46–61. [Google Scholar] [CrossRef] [Green Version]
- Zininga, T.; Shonhai, A. Are heat shock proteins druggable candidates? Am. J. Biochem. Biotechnol. 2014, 10, 208–210. [Google Scholar] [CrossRef] [Green Version]
- Njunge, J.M.; Mandal, P.; Przyborski, J.M.; Boshoff, A.; Pesce, E.-R.; Blatch, G.L. PFB0595w is a Plasmodium falciparum J protein that co-localizes with PfHsp70-1 and can stimulate its in vitro ATP hydrolysis activity. Int. J. Biochem. Cell Biol. 2015, 62, 47–53. [Google Scholar] [CrossRef]
- Bascos, N.A.D.; Landry, S.J. Structural rigidity regulates functional interactions in the Hsp40-Hsp70 molecular machine. Biophys. J. 2015, 108, 210a. [Google Scholar] [CrossRef] [Green Version]
- Nyakundi, D.O.; Bentley, S.J.; Boshoff, A. Hsp70 escort protein: More than a regulator of mitochondrial Hsp70. Curr. Proteom. 2019, 16, 64–73. [Google Scholar] [CrossRef]
- LaCount, D.J.; Vignali, M.; Chettier, R.; Phansalkar, A.; Bell, R.; Hesselberth, J.R.; Schoenfeld, L.W.; Ota, I.; Sahasrabudhe, S.; Kurschner, C.; et al. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature 2005, 438, 103–107. [Google Scholar] [CrossRef]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef]
- Raviol, H.; Bukau, B.; Mayer, M.P. Human and yeast Hsp110 chaperones exhibit functional differences. FEBS Lett. 2005, 580, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Sarbeng, E.B.; Vorvis, C.; Kumar, D.P.; Zhou, L.; Liu, Q. Unique peptide substrate binding properties of 110-kDa Heat-shock protein (Hsp110) determine its distinct chaperone activity. J. Biol. Chem. 2012, 287, 5661–5672. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.J.; Eston, D.; Murawski, M.; Kaneneko, Y.; Subjeck, J.R. The chaperoning activity of Hsp110: Identification of functional domains by use of targeted deletions. J. Biol. Chem. 1999, 27, 15712–15718. [Google Scholar] [CrossRef] [Green Version]
- Polier, S.; Hartl, F.U.; Bracher, A. Interaction of the Hsp110 molecular chaperones from S. cerevisiae with substrate protein. J. Mol. Biol. 2010, 401, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Goeckeler, J.L.; Stephens, A.; Lee, P.; Caplan, A.J.; Brodsky, J.L. Overexpression of yeast Hsp110 homolog Sse1p suppresses ydj1-151 thermosensitivity and restores Hsp90-dependent activity. Mol. Biol. Cell 2002, 13, 2760–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadlish, H.; Rampelt, H.; Shorter, J.; Wegrzyn, R.D.; Andréasson, C.; Lindquist, S.; Bukau, B. Hsp110 chaperones regulate prion formation and propagation in S. cerevisiae by two discrete activities. PLoS ONE 2008, 3, e0001763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packschies, L.; Theyssen, H.; Buchberger, A.; Bukau, B.; Goody, R.S.; Reinstein, J. GrpE accelerates nucleotide exchange of the molecular chaperone DnaK with an associative displacement mechanism. Biochemistry 1997, 36, 3417–3422. [Google Scholar] [CrossRef] [PubMed]
- Bimston, D.; Song, J.; Winchester, D.; Takayama, S.; Reed, J.C.; Morimoto, R.I. BAG-1, a negative regulator of Hsp70 chaperone activity, uncouples nucleotide hydrolysis from substrate release. EMBO J. 1998, 17, 6871–6878. [Google Scholar] [CrossRef] [Green Version]
- Patury, S.; Miyata, Y.; Gestwicki, J.E. Pharmacological targeting of the Hsp70 Chaperone. Curr. Top. Med. Chem. 2009, 9, 1337–1351. [Google Scholar] [CrossRef] [Green Version]
- Mandal, A.K.; Gibney, P.A.; Nillegoda, N.B.; Theodoraki, M.A.; Caplan, A.J.; Morano, K.A. Hsp110 chaperones control client fate determination in the Hsp70-Hsp90 chaperone system. Mol. Biol. Cell 2010, 21, 1439–2448. [Google Scholar] [CrossRef] [Green Version]
- Muralidharan, V.; Oksman, A.; Pal, P.; Lindquist, S.; Goldberg, D.E. Plasmodium falciparum heat shock protein 110 stabilizes the asparagine repeat-rich parasite proteome during malarial fevers. Nat. Commun. 2012, 3, 1310. [Google Scholar] [CrossRef] [Green Version]
- Akide-Ndunge, O.B.; Tambini, E.; Giribaldi, G.; McMillan, P.J.; Müller, S.; Arese, P.; Turrini, F. Co-ordinated stage-dependent enhancement of Plasmodium falciparum antioxidant enzymes and heat shock protein expression in parasites growing in oxidatively stressed or G6PD-deficient red blood cells. Malar. J. 2009, 8, 113. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, C.L.; Rosenthal, A.S.; D’Angelo, J.; Griffin, C.E.; Posner, G.H.; Cooper, R.A. Accumulation of artemisinin trioxane derivatives within neutral lipids of Plasmodium falciparum malaria parasites is endoperoxide-dependent. Biochem. Pharmacol. 2009, 77, 322–336. [Google Scholar] [CrossRef] [Green Version]
- Bhartiya, D.; Chawla, V.; Ghosh, S.; Shankar, R.; Kumar, N. Genome-wide regulatory dynamics of G-quadruplexes in human malaria parasite Plasmodium falciparum. Genomics 2016, 108, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, H.K.; Koay, Y.C.; Irani, S.; Das, R.; Nassar, Z.D.; Australian Prostate Cancer BioResource; Butler, L.M. A novel class of Hsp90 C-terminal modulators have pre-clinical efficacy in prostate tumor cells without induction of a heat shock response. Prostate 2016, 76, 1546–1559. [Google Scholar] [CrossRef] [PubMed]
- Edkins, A.L.; Price, J.T.; Pockley, A.G.; Blatch, G.L. Heat shock proteins as modulators and therapeutic targets of chronic disease: An integrated perspective. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 73, 20160521. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zininga, T.; Shonhai, A. Small Molecule Inhibitors Targeting the Heat Shock Protein System of Human Obligate Protozoan Parasites. Int. J. Mol. Sci. 2019, 20, 5930. https://doi.org/10.3390/ijms20235930
Zininga T, Shonhai A. Small Molecule Inhibitors Targeting the Heat Shock Protein System of Human Obligate Protozoan Parasites. International Journal of Molecular Sciences. 2019; 20(23):5930. https://doi.org/10.3390/ijms20235930
Chicago/Turabian StyleZininga, Tawanda, and Addmore Shonhai. 2019. "Small Molecule Inhibitors Targeting the Heat Shock Protein System of Human Obligate Protozoan Parasites" International Journal of Molecular Sciences 20, no. 23: 5930. https://doi.org/10.3390/ijms20235930
APA StyleZininga, T., & Shonhai, A. (2019). Small Molecule Inhibitors Targeting the Heat Shock Protein System of Human Obligate Protozoan Parasites. International Journal of Molecular Sciences, 20(23), 5930. https://doi.org/10.3390/ijms20235930