Role of the Novel Hsp90 Co-Chaperones in Dynein Arms’ Preassembly

Abstract
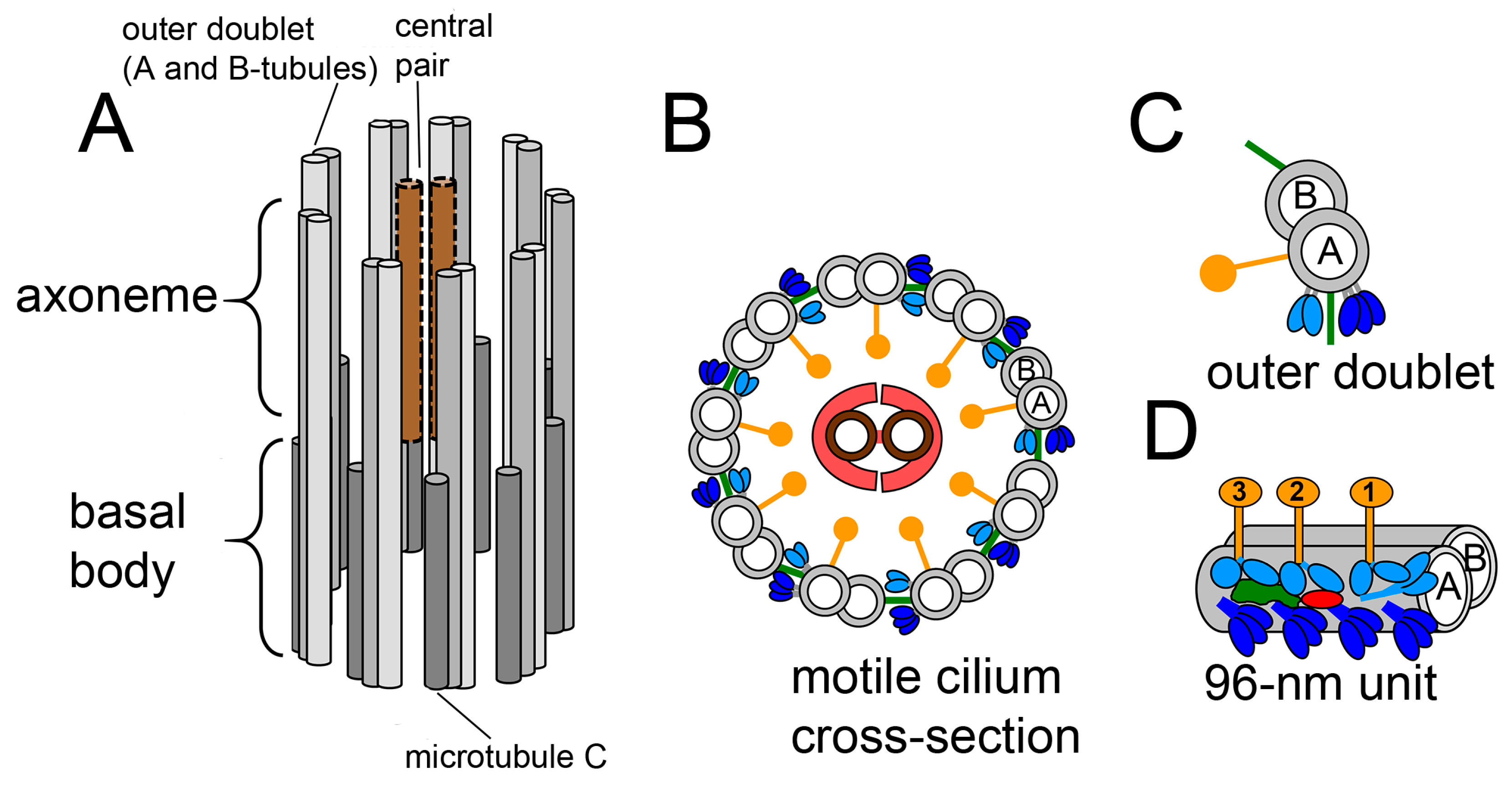
:1. Introduction
2. Hsp90 and Its Co-Chaperones
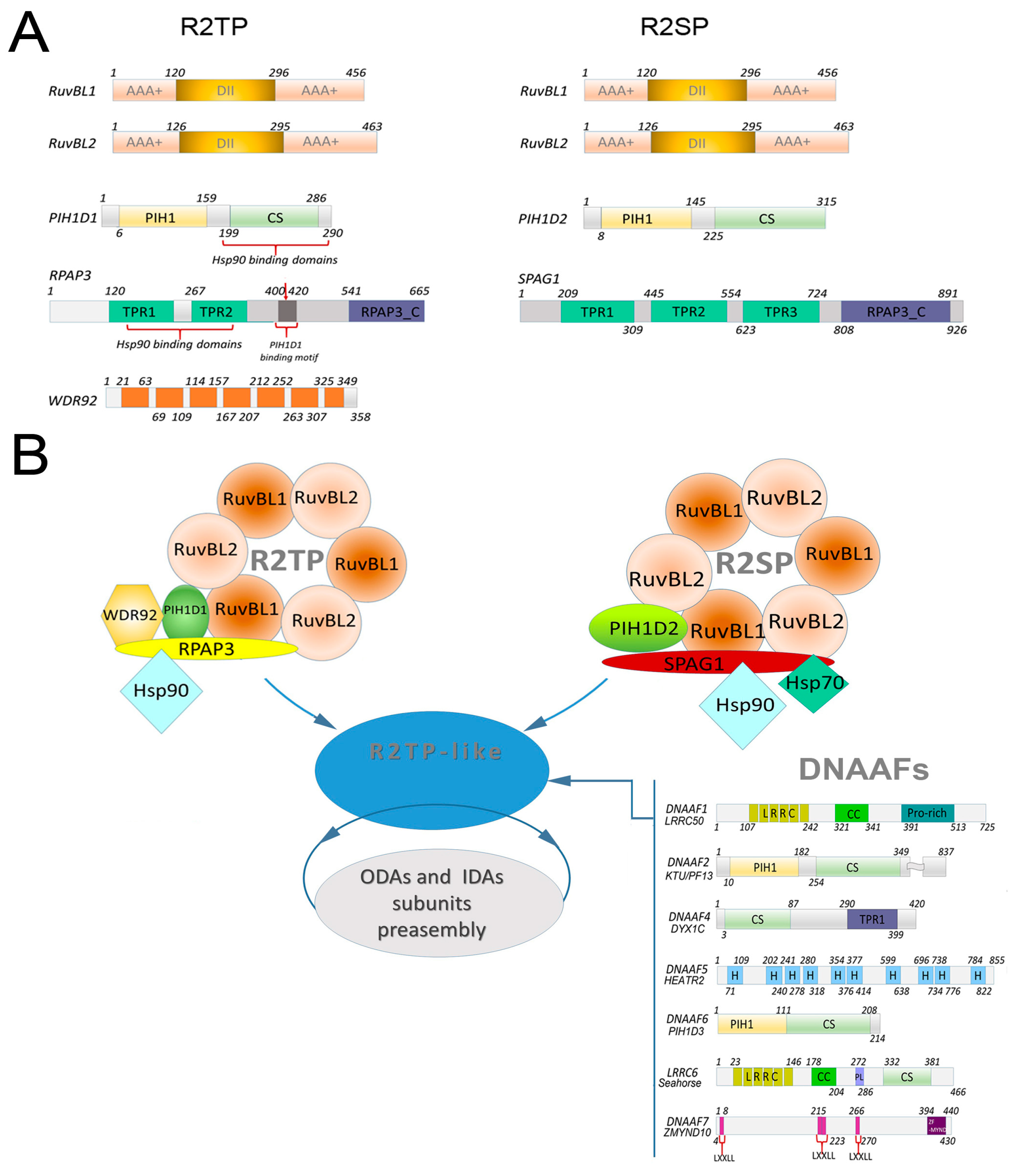
2.1. Composition of R2TP Complex
2.2. R2TP Complexes in the Dynein Arm Assembly
2.3. Composition of R2SP Complex and Function in Dynein Arm Preassembly
2.4. DNAAFs and Their Function in Dynein Arm Assembly
3. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pitnick, S.; Markow, T.A.; Spicer, G.S. Delayed male maturity is a cost of producing large sperm in Drosophila. Proc. Natl. Acad. Sci. USA 1995, 92, 10614–10618. [Google Scholar] [CrossRef] [Green Version]
- Fliegauf, M.; Benzing, T.; Omran, H. When cilia go bad: Cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007, 8, 880–893. [Google Scholar] [CrossRef]
- Nonaka, S.; Shiratori, H.; Saijoh, Y.; Hamada, H. Determination of left-right patterning of the mouse embryo by artificial nodal flow. Nature 2002, 418, 96–99. [Google Scholar] [CrossRef]
- Ishikawa, H.; Marshall, W.F. Ciliogenesis: Building the cell’s antenna. Nat. Rev. Mol. Cell Biol. 2011, 12, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Tanaka, Y.; Okada, Y. Left-right determination: Involvement of molecular motor KIF3, cilia, and nodal flow. Cold Spring Harb. Perspect. Biol. 2009, 1, a000802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicastro, D.; Schwartz, C.; Pierson, J.; Gaudette, R.; Porter, M.E.; Mcintosh, J.R. The molecular architecture of axonemes revealed by cryoelectron tomography. Science 2006, 313, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, P.; Joachimiak, E.; Bazan, R.; Fabczak, H.; Włoga, D. Motile cilia—From ultrastructure to function. Kosmos 2018, 67, 195–205. [Google Scholar]
- Kamiya, R. Functional diversity of axonemal dyneins as studied in Chlamydomonas mutants. Int. Rev. Cytol. 2002, 219, 115–155. [Google Scholar] [CrossRef]
- Kollmar, M. Fine-Tuning Motile Cilia and Flagella: Evolution of the dynein motor proteins from plants to humans at high resolution. Mol. Biol. Evol. 2016, 33, 3249–3267. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, R.; Yagi, T. Functional diversity of axonemal dyneins as assessed by in vitro and in vivo motility assays of Chlamydomonas mutants. Zoolog. Sci. 2014, 31, 633–644. [Google Scholar] [CrossRef]
- Ishikawa, T. Axoneme Structure from Motile Cilia. Cold Spring Harb. Perspect. Biol. 2017, 9, a028076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fok, A.K.; Wang, H.; Katayama, A.; Aihara, M.S.; Allen, R.D. 22S axonemal dynein is preassembled and functional prior to being transported to and attached on the axonemes. Cell Motil. Cytoskelet. 1994, 29, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Fowkes, M.E.; Mitchell, D.R. The role of preassembled cytoplasmic complexes in assembly of flagellar dynein subunits. Mol. Biol. Cell 1998, 9, 2337–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.T.; Gao, C.; Lucker, B.F.; Cole, D.G.; Mitchell, D.R. ODA16 aids axonemal outer row dynein assembly through an interaction with the intraflagellar transport machinery. J Cell Biol. 2008, 183, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhao, L.; Yuan, S.; Zhang, J.; Sun, Z. Axonemal dynein assembly requires the R2TP complex component Pontin. Development 2017, 144, 4684–4693. [Google Scholar] [CrossRef] [Green Version]
- Omran, H.; Kobayashi, D.; Olbrich, H.; Tsukahara, T.; Loges, N.T.; Hagiwara, H.; Zhang, Q.; Leblond, G.; O’Toole, E.; Hara, C.; et al. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature 2008, 456, 611–616. [Google Scholar] [CrossRef] [Green Version]
- Mitchison, H.M.; Schmidts, M.; Loges, N.T.; Freshour, J.; Dritsoula, A.; Hirst, R.A.; O’Callaghan, C.; Blau, H.; Al Dabbagh, M.; Olbrich, H.; et al. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012, 44, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.B.; Dean, A.B.; Mitchell, D.R. Cytoplasmic preassembly and trafficking of axonemal dyneins. In The Biology of Dynein Motors; King, S., Ed.; Elsevier Inc.: Cambridge, MA, USA, 2018; pp. 141–161. [Google Scholar] [CrossRef]
- Kakihara, Y.; Houry, W.A. The R2TP complex: Discovery and functions. Biochim. Biophys. Acta 2012, 1823, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Li, J.; Buchner, J. Structure, function and regulation of the Hsp90 machinery. Biomed. J. 2013, 36, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Lynham, J.; Houry, W.A. The Multiple Functions of the PAQosome: An R2TP- and URI1 Prefoldin-Based Chaperone Complex. Adv. Exp. Med. Biol. 2018, 1106, 37–72. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Davey, M.; Hsu, Y.C.; Kaplanek, P.; Tong, A.; Parsons, A.B.; Krogan, N.; Cagney, G.; Mai, D.; Greenblatt, J.; et al. Navigating the chaperone network: An integrative map of physical and genetic interactions mediated by the Hsp90 chaperone. Cell 2005, 120, 715–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Hernández, H.; Pal, M.; Rodríguez, C.F.; Fernandez-Leiro, R.; Prodromou, C.; Pearl, L.H.; Llorca, O. Structural mechanism for regulation of the AAA-ATPases RUVBL1-RUVBL2 in the R2TP co-chaperone revealed by cryo-EM. Sci. Adv. 2019, 5, eaaw1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Hernández, H.; Pal, M.; Rodríguez, C.F.; Prodromou, C.; Pearl, L.H.; Llorca, O. Advances on the structure of the R2TP/Prefoldin-like complex. Adv. Exp. Med. Biol. 2018, 1106, 73–83. [Google Scholar] [CrossRef]
- Huizar, R.L.; Lee, C.; Boulgakov, A.A.; Horani, A.; Tu, F.; Marcotte, E.M.; Brody, S.L.; Wallingford, J.B. A liquid-like organelle at the root of motile ciliopathy. eLife 2018, 7, e38497. [Google Scholar] [CrossRef]
- Knowles, M.R.; Ostrowski, L.E.; Loges, N.T.; Hurd, T.; Leigh, M.W.; Huang, L.; Wolf, W.E.; Carson, J.L.; Hazucha, M.J.; Yin, W.; et al. Mutations in SPAG1 cause primary ciliary dyskinesia associated with defective outer and inner dynein arms. Am. J. Hum. Genet. 2013, 93, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Duquesnoy, P.; Escudier, E.; Vincensini, L.; Freshour, J.; Bridoux, A.M.; Coste, A.; Deschildre, A.; de Blic, J.; Legendre, M.; Montantin, G.; et al. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2009, 85, 890–896. [Google Scholar] [CrossRef] [Green Version]
- Tarkar, A.; Loges, N.T.; Slagle, C.E.; Francis, R.; Dougherty, G.W.; Tamayo, J.V.; Shook, B.; Cantino, M.; Schwartz, D.; Jahnke, C.; et al. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet. 2013, 45, 995–1003. [Google Scholar] [CrossRef]
- Horani, A.; Druley, T.E.; Zariwala, M.A.; Patel, A.C.; Levinson, B.T.; Van Arendonk, L.G.; Thornton, K.C.; Giacalone, J.C.; Albee, A.J.; Wilson, K.S.; et al. Whole-exome capture and sequencing identifies HEATR2mutation as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2012, 91, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Diggle, C.P.; Moore, D.J.; Mali, G.; zur Lage, P.; Ait-Lounis, A.; Schmidts, M.; Shoemark, A.; Garcia Munoz, A.; Halachev, M.R.; Gautier, P.; et al. HEATR2 plays a conserved role in assembly of the ciliary motile apparatus. PLoS Genet. 2014, 10, e1004577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paff, T.; Loges, N.T.; Aprea, I.; Wu, K.; Bakey, Z.; Haarman, E.G.; Daniels, J.M.A.; Sistermans, E.A.; Bogunovic, N.; Dougherty, G.W.; et al. Mutations in PIH1D3 cause X-linked primary ciliary dyskinesia with outer and inner dynein arm defects. Am. J. Hum. Genet. 2017, 100, 160–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olcese, C.; Patel, M.P.; Shoemark, A.; Kiviluoto, S.; Legendre, M.; Williams, H.J.; Vaughan, C.K.; Hayward, J.; Goldenberg, A.; Emes, R.D.; et al. X-linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3. Nat. Commun. 2017, 8, 14279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zariwala, M.A.; Gee, H.Y.; Kurkowiak, M.; Al-Mutairi, D.A.; Leigh, M.W.; Hurd, T.W.; Hjeij, R.; Dell, S.D.; Chaki, M.; Dougherty, G.W.; et al. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am. J. Hum. Genet. 2013, 93, 336–345. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.J.; Onoufriadis, A.; Shoemark, A.; Simpson, M.A.; zur Lage, P.I.; de Castro, S.C.; Bartoloni, L.; Gallone, G.; Petridi, S.; Woollard, W.J.; et al. Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 93, 346–356. [Google Scholar] [CrossRef]
- Kott, E.; Duquesnoy, P.; Copin, B.; Legendre, M.; Dastot-Le Moal, F.; Montantin, G.; Jeanson, L.; Tamalet, A.; Papon, J.F.; Siffroi, J.P.; et al. Loss-of-function mutations in lrrc6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2012, 91, 958–964. [Google Scholar] [CrossRef] [Green Version]
- Horani, A.; Ferkol, T.W.; Shoseyov, D.; Wasserman, M.G.; Oren, Y.S.; Kerem, B.; Amirav, I.; Cohen-Cymberknoh, M.; Dutcher, S.K.; Brody, S.L.; et al. LRRC6 mutation causes primary ciliary dyskinesia with dynein arm defects. PLoS ONE 2013, 8, e59436. [Google Scholar] [CrossRef]
- Inaba, Y.; Shinohara, K.; Botilde, Y.; Nabeshima, R.; Takaoka, K.; Ajima, R.; Lamri, L.; Takeda, H.; Saga, Y.; Nakamura, T.; et al. Transport of the outer dynein arm complex to cilia requires a cytoplasmic protein Lrrc6. Genes Cells 2016, 21, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Pal, M.; Morgan, M.; Phelps, S.E.; Roe, S.M.; Parry-Morris, S.; Downs, J.A.; Polier, S.; Pearl, L.H.; Prodromou, C. Structural basis for phosphorylation-dependent recruitment of Tel2 to Hsp90 by Pih1. Structure 2014, 22, 805–818. [Google Scholar] [CrossRef] [Green Version]
- Horejsi, Z.; Takai, H.; Adelman, C.A.; Collis, S.J.; Flynn, H.; Maslen, S.; Skehel, J.M.; de Lange, T.; Boulton, S.J. CK2 phospho-dependent binding of R2TP complex to TEL2 is essential for mTOR and SMG1 stability. Mol. Cell. 2010, 39, 839–850. [Google Scholar] [CrossRef] [Green Version]
- Horejsi, Z.; Stach, L.; Flower, T.G.; Joshi, D.; Flynn, H.; Skehel, J.M.; O’Reilly, N.J.; Ogrodowicz, R.W.; Smerdon, S.J.; Boulton, S.J. Phosphorylation-dependent PIH1D1 interactions define substrate specificity of the R2TP cochaperone complex. Cell Rep. 2014, 7, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.M.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benbahouche, N.H.; Iliopoulos, I.; Török, I.; Marhold, J.; Henri, J.; Kajava, A.V.; Farkaš, R.; Kempf, T.; Schnölzer, M.; Meyer, P.; et al. Drosophila Spag is the homolog of RNA polymerase II-associated protein 3 (RPAP3) and recruits the heat shock proteins 70 and 90 (Hsp70 and Hsp90) during the assembly of cellular machineries. J. Biol. Chem. 2014, 289, 6236–6247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henri, J.; Chagot, M.E.; Bourguet, M.; Abel, Y.; Terral, G.; Maurizy, C.; Aigueperse, C.; Georgescauld, F.; Vandermoere, F.; Saint-Fort, R.; et al. Deep structural analysis of RPAP3 and PIH1D1, two components of the HSP90 co-chaperone R2TP complex. Structure 2018, 26, 1196–1209. [Google Scholar] [CrossRef] [Green Version]
- Martino, F.; Pal, M.; Muñoz-Hernández, H.; Rodríguez, C.F.; Núñez-Ramírez, R.; Gil-Carton, D.; Degliesposti, G.; Skehel, J.M.; Roe, S.M.; Prodromou, C.; et al. RPAP3 provides a flexible scaffold for coupling HSP90 to the human R2TP co-chaperone complex. Nat. Commun. 2018, 9, 1501. [Google Scholar] [CrossRef] [Green Version]
- Stolc, V.; Samanta, M.P.; Tongprasit, W.; Marshall, W.F. Genome-wide transcriptional analysis of flagellar regeneration in Chlamydomonas reinhardtii identifies orthologs of ciliary disease genes. Proc. Natl. Acad. Sci. USA 2005, 102, 3703–3707. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Yuan, S.; Cao, Y.; Kallakuri, S.; Li, Y.; Kishimoto, N.; DiBella, L.; Sun, Z. Reptin/Ruvbl2 is a Lrrc6/Seahorse interactor essential for cilia motility. Proc. Natl. Acad. Sci. USA 2013, 110, 12697–12702. [Google Scholar] [CrossRef] [Green Version]
- Dafinger, C.; Rinschen, M.M.; Borgal, L.; Ehrenberg, C.; Basten, S.G.; Franke, M.; Höhne, M.; Rauh, M.; Göbel, H.; Bloch, W.; et al. Targeted deletion of the AAA-ATPase Ruvbl1 in mice disrupts ciliary integrity and causes renal disease and hydrocephalus. Exp. Mol. Med. 2018, 50, 75. [Google Scholar] [CrossRef] [Green Version]
- Tammana, D.; Tammana, T.V.S. Human DNA helicase, RuvBL1 and its Chlamydomonas homologue, CrRuvBL1 plays an important role in ciliogenesis. Cytoskeleton 2017, 74, 251–259. [Google Scholar] [CrossRef]
- Yamamoto, R.; Hirono, M.; Kamiya, R. Discrete PIH proteins function in the cytoplasmic preassembly of different subsets of axonemal dyneins. J. Cell Biol. 2010, 190, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Oda, T.; Kikkawa, M.; Takeda, H. Systematic studies of all PIH proteins in zebrafish reveal their distinct roles in axonemal dynein assembly. eLife 2018, 7, e36979. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wang, L.; Pan, J. Chlamydomonas WDR92 in association with R2TP-like complex and multiple DNAAFs to regulate ciliary dynein preassembly. J. Mol. Cell Biol. 2019, 11, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Patel-King, R.S.; Sakato-Antoku, M.; Yankova, M.; King, S.M. WDR92 is required for axonemal dynein heavy chain stability in cytoplasm. Mol. Biol. Cell. 2019, 30, 1834–1845. [Google Scholar] [CrossRef] [PubMed]
- Zur-Lage, P.; Stefanopoulou, P.; Styczynska-Soczka, K.; Quinn, N.; Mali, G.; von Kriegsheim, A.; Mill, P.; Jarman, A.P. Ciliary dynein motor preassembly is regulated by Wdr92 in association with HSP90 co-chaperone, R2TP. J. Cell Biol. 2018, 217, 2583–2598. [Google Scholar] [CrossRef] [Green Version]
- Hartill, V.L.; van de Hoek, G.; Patel, R.; Little, M.P.; Watson, C.M.; Berry, I.R.; Shoemark, A.; Abdelmottaleb, D.; Parkes, E.; Bacchelli, C.; et al. DNAAF1 links HEATR laterality with the AAA+ ATPase RUVBL1 and ciliary intraflagellar transport. Hum. Mol. Genet. 2018, 27, 529–545. [Google Scholar] [CrossRef]
- Loges, N.T.; Olbrich, H.; Becker-Heck, A.; Haffner, K.; Heer, A.; Reinhard, C.; Schmidts, M.; Kispert, A.; Zariwala, M.A.; Leigh, M.W.; et al. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009, 85, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.J.; Noh, S.H.; Han, S.M.; Choi, W.I.; Kim, H.Y.; Yu, S.; Lee, J.S.; Rim, J.H.; Lee, M.G.; Hildebrandt, F.; et al. ZMYND10 stabilizes intermediate chain proteins in the cytoplasmic pre-assembly of dynein arms. PLoS Genet. 2018, 14, e1007316. [Google Scholar] [CrossRef] [Green Version]
- Boulon, S.; Pradet-Balade, B.; Verheggen, C.; Molle, D.; Boireau, S.; Georgieva, M.; Azzag, K.; Robert, M.C.; Ahmad, Y.; Neel, H.; et al. HSP90 and its R2TP/Prefoldin-like cochaperone are involved in the cytoplasmic assembly of RNA polymerase II. Mol. Cell 2010, 39, 912–924. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Saeki, M.; Egusa, H.; Niwa, H.; Kamisaki, Y. PIH1D1, a subunit of R2TP complex, inhibits doxorubicin-induced apoptosis. Biochem. Biophys. Res. Commun. 2010, 403, 340–344. [Google Scholar] [CrossRef]
- Patel-King, R.S.; King, S.M. A prefoldin-associated WD-repeatprotein (WDR92) is required for the correct architectural assembly of motile cilia. Mol. Biol. Cell 2016, 27, 1204–1209. [Google Scholar] [CrossRef]
- Itsuki, Y.; Saeki, M.; Nakahara, H.; Egusa, H.; Irie, Y.; Terao, Y.; Kawabata, S.; Yatani, H.; Kamisaki, Y. Molecular cloning of novel Monad binding protein containing tetratricopeptide repeat domains. FEBS Lett. 2008, 582, 2365–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurizy, C.; Quinternet, M.; Abel, Y.; Verheggen, C.; Santo, P.E.; Bourguet, M.; Paiva, A.C.F.; Bragantini, B.; Chagot, M.E.; Robert, M.C.; et al. The RPAP3-Cterminal domain identifies R2TP-like quaternary chaperones. Nat. Commun. 2018, 9, 2093. [Google Scholar] [CrossRef] [PubMed]
- Chagot, M.E.; Dos Santos Morais, R.; Dermouche, S.; Lefebvre, D.; Manival, X.; Chipot, C.; Dehez, F.; Quinternet, M. Binding properties of the quaternary assembly protein SPAG1. Biochem. J. 2019, 476, 1679–1694. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Ustione, A.; Huang, T.; Firth, A.L.; Pan, J.; Gunsten, S.P.; Haspel, J.A.; Piston, D.W.; Brody, S.L. Establishment of the early cilia preassembly protein complex during motile ciliogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E1221–E1228. [Google Scholar] [CrossRef] [Green Version]
- Cloutier, P.; Poitras, C.; Durand, M.; Hekmat, O.; Fiola-Masson, É.; Bouchard, A.; Faubert, D.; Chabot, B.; Coulombe, B. R2TP/Prefoldin-like component RUVBL1/RUVBL2 directly interacts with ZNHIT2 to regulate assembly of U5 small nuclear ribonucleoprotein. Nat. Commun. 2017, 8, 15615. [Google Scholar] [CrossRef] [Green Version]
- Dong, F.; Shinohara, K.; Botilde, Y.; Nabeshima, R.; Asai, Y.; Fukumoto, A.; Hasegawa, T.; Matsuo, M.; Takeda, H.; Shiratori, H.; et al. Pih1d3 is required for cytoplasmic preassembly of axonemal dynein in mouse sperm. J. Cell Biol. 2014, 204, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhao, M.; Wang, S.; Chen, J.; Wang, Y.; Cao, Q.; Zhou, W.; Liu, J.; Xu, Z.; Tong, G.; et al. A novel role for DYX1C1, a chaperone protein for both Hsp70 and Hsp90, in breast cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 1265–1276. [Google Scholar] [CrossRef]
- Miao, C.; Jiang, Q.; Li, H.; Zhang, Q.; Bai, B.; Bao, Y.; Zhang, T. Mutations in the motile cilia gene DNAAF1 are associated with neural tube defects in humans. G3 2016, 6, 3307–3316. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, W.; Gaudet, J.; Cheney, M.D.; Roudaia, L.; Cierpicki, T.; Klet, R.C.; Hartman, K.; Laue, T.M.; Speck, N.A.; et al. Structural basis for recognition of SMRT/N-CoR by the MYND domain and its contribution to AML1/ETO’s activity. Cancer Cell 2007, 11, 483–497. [Google Scholar] [CrossRef] [Green Version]
- McClintock, T.S.; Glasser, C.E.; Bose, S.C.; Bergman, D.A. Tissue expression patterns identify mouse cilia genes. Physiol. Genom. 2008, 32, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, D.; Asano-Hoshino, A.; Nakakura, T.; Nishimaki, T.; Ansai, S.; Kinoshita, M.; Ogawa, M.; Hagiwara, H.; Yokoyama, T. Loss of zinc finger MYND-type containing 10 (zmynd10) affects cilia integrity and axonemal localization of dynein arms, resulting in ciliary dysmotility, polycystic kidney and scoliosis in medaka (Oryzias latipes). Dev. Biol. 2017, 430, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Kurkowiak, M.; Ziętkiewicz, E.; Greber, A.; Voelkel, K.; Wojda, A.; Pogorzelski, A.; Witt, M. ZMYND10-mutation analysis in slavic patients with primary ciliary dyskinesia. PLoS ONE 2016, 11, e0148067. [Google Scholar] [CrossRef] [PubMed]
- Mali, G.R.; Yeyati, P.L.; Mizuno, S.; Dodd, D.O.; Tennant, P.A.; Keighren, M.A.; Zur Lage, P.; Shoemark, A.; Garcia-Munoz, A.; Shimada, A.; et al. ZMYND10 functions in a chaperone relay during axonemal dynein assembly. eLife 2018, 7, e34389. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Chen, W.; Huang, J.; Wang, L.; Qian, L. Clinical and genetic analysis of patients with primary ciliary dyskinesia caused by novel DNAAF3 mutations. J. Hum. Genet. 2019, 64, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Fassad, M.R.; Shoemark, A.; le Borgne, P.; Koll, F.; Patel, M.; Dixon, M.; Hayward, J.; Richardson, C.; Frost, E.; Jenkins, L.; et al. C11orf70 mutations disrupting the intraflagellar transport-dependent assembly of multiple axonemal dyneins cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2018, 102, 956–972. [Google Scholar] [CrossRef] [Green Version]
- Höben, I.M.; Hjeij, R.; Olbrich, H.; Dougherty, G.W.; Nöthe-Menchen, T.; Aprea, I.; Frank, D.; Pennekamp, P.; Dworniczak, B.; Wallmeier, J.; et al. Mutations in C11orf70 cause primary ciliary dyskinesia with randomization of left/right body asymmetry due to defects of outer and inner dynein arms. Am. J. Hum. Genet. 2018, 102, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Zietkiewicz, E.; Bukowy-Bieryllo, Z.; Rabiasz, A.; Daca-Roszak, P.; Wojda, A.; Voelkel, K.; Rutkiewicz, E.; Pogorzelski, A.; Rasteiro, M.; Witt, M. CFAP300: Mutations in Slavic patients with primary ciliary dyskinesia and a role in ciliary dynein arms trafficking. Am. J. Respir. Cell Mol. Biol. 2019, 61, 440–449. [Google Scholar] [CrossRef]
- Austin-Tse, C.; Halbritter, J.; Zariwala, M.A.; Gilberti, R.M.; Gee, H.Y.; Hellman, N.; Pathak, N.; Liu, Y.; Panizzi, J.R.; Patel-King, R.S.; et al. Zebrafish ciliopathy screen plus human mutational analysis identifies C21orf59 and CCDC65 defects as causing primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 93, 672–686. [Google Scholar] [CrossRef] [Green Version]
- Lucas, J.S.; Burgess, A.; Mitchison, H.M.; Moya, E.; Williamson, M.; Hogg, C.; National PCD Service, UK. Diagnosis and management of primary ciliary dyskinesia. Arch. Dis. Child. 2014, 99, 850–856. [Google Scholar] [CrossRef] [Green Version]
- Boon, M.; Wallmeier, J.; Ma, L.; Loges, N.T.; Jaspers, M.; Olbrich, H.; Dougherty, G.W.; Raidt, J.; Werner, C.; Amirav, I.; et al. MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat. Commun. 2014, 5, 4418. [Google Scholar] [CrossRef]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Zariwala, M.; Leigh, M. Primary Ciliary Dyskinesia. Clin. Chest. Med. 2016, 37, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchison, H.M.; Valente, E.M. Motile and non-motile cilia in human pathology: From function to phenotypes. J. Pathol. 2017, 241, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Tanaka, Y.; Okada, Y. Cilia, KIF3 molecular motor and nodal flow. Curr. Opin. Cell Biol. 2012, 24, 31–39. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Human Protein | Aliases | Protein Accession Number | Protein Size (kDa) | Localization in Cell | Domains | * Chaperone, Co-Chaprone and DNAAFs Interaction | PCD |
|---|---|---|---|---|---|---|---|
| RuvBL1, | Pontin | Q9Y265 | 50 | DynAPs | Hsp90, RuvBL2, PIH1D1, RPAP3, WDR92, SPAG1 | _ | |
| RuvBL2 | Reptin | Q9Y230 | 51 | DynAPs | RuvBL1, PIH1D1, RPAP3, WDR92 | _ | |
| RPAP3 | HSpagh | Q9H6T3 | 75 | _ | TPR- RPAP3-C-ter | Hsp70, Hsp90, RuvBL1, RuvBL2, PIH1D1, WDR92, | |
| PIH1D1 | Q9NWS0 | 32 | _ | PIH1, CS | Hsp90, RuvBL1, RuvBL2; RPAP3, WDR92, DNAAF4 | _ | |
| PIH1D2 | Q8WWB5 | 39.4 | _ | PIH1, CS | RuvBL1; RuvBL2, SPAG1 | _ | |
| WDR92 | Monad | Q96MX6 | 39.7 | DynAPs | WD40 | RuvBL1, RuvBL2; RPAP3, PIH1D1, DNAAF1, DNAAF4, SPAG1 | |
| SPAG1 | Q07617 | 103.6 | DynAPs | TPR, RPAP3-C-ter | Hsp70, Hsp90, RuvBL1, RuvBL2, PIH1D2, WDR92, DNAAF2, DNAAF4, DNAAF5, DNAAF6 | MIM:615505 [28] | |
| DNAAF1 | LRRC50/ ODA7 | Q8NEP3 | 40 | cytoplasm | LRR | Hsp90, RuvBL1, RuvBL2, | MIM:613193 [29] |
| DNAAF2 | KTU/ PF13 | Q9NVR5 | 91 | DynAPs | PIH1, CS | HSP70, HSP90, RuvBL1, RuvBL2, DNAAF4 | MIM:612518 [16] |
| DNAAF4 | DYX1C1 | Q86X45 | 48.5 | DynAPs | TPR, CS | Hsp70, Hsp90, DNAAF4 RuvBL1, RuvBL2 DNAAF2 | MIM:615482 [30] |
| DNAAF5 | HEATR2 | Q86Y56 | 93.5 | DynAPs | HEAT_type_2 | DNAAF2, SPAG1, | MIM:614874 [31,32] |
| DNAAF6 | PIH1D3 | Q9NQM4 | 24 | DynAPs | PIH1, CS | Hsp70, Hsp90, DNAAF1, DNAAF2, DNAAF3 | CILD36 [33,34] |
| DNAAF7 | ZMYND10 | O75800 | 50 | DynAPs | Znf_MYND | Hsp90, Hsc70 RuvBL2, LRRC6, DNAAF4, | MIM:615444 [35,36] |
| LRRC6 | Seahorse | Q86X45 | 54 | DynAPs | LRR, CS | RuvBL1, RuvBL2 DNAAF4, | MIM:614935 [37,38,39] |
| Chlamydomonas | Danio rerio | Mouse | Humans | |||||
|---|---|---|---|---|---|---|---|---|
| ODA | IDA | ODA | IDA | ODA | IDA | ODA | IDA | |
| RuvBL1-RuvBL2 | DNAH9, DNAI2 [15,49] | DNALI1 [49] | ||||||
| PIH1D1/MOT48 | IDA b, c, [51] | DNAI1 [52] | IDAc [52] | |||||
| PIH1D2 | DNAH8, DNAI1 [52] | IDA b,c,e [52] | ||||||
| WDR92 | αHC, βHC, ϒHC, IC/LC [53,54] | DHC5, DHC8, DHC9 [53] | DNAH17, DNAH8 DNAI1-DNAI2, DNAL4 [55] | DNAH12, DNAH10, Centrin [55] | ||||
| SPAG1 | DNAH5, DNAI1 [28] | DNALI1 [28] | ||||||
| DNAAF1/ODA7 | HCα, IC70, IC78 [13] | ID1 [13] | DNAH5, DNAH9, DNAI2 [56,57] | DNALI1 [56,57] | ||||
| DNAAF2 | HCα, HCβ, HCϒ, [16] | HC9 [16] | DNAH8 DNAI1 [52] | IDA b,c,e [52] | DNAH5, DNAH9, DNAI2 [16] | DNALI1 [16] | ||
| DNAAF4 | DNAH5, DNAH9, DNAI2 [30] | DNALI1 [30] | DNAH5 [30] | DNALI1 [30] | ||||
| DNAAF5 | DIC2/IC78 [32] | DNALI1 [31] | DNAH5 [31] | DNALI1 [31] | ||||
| DNAAF6 | DNAH8, DNAI1 [53] | IDA c,d,g [52] | DNAH5, DNAI1, [34] | DNALI1 [34] | ||||
| DNAAF7 | DNAH5, DNAI2 [58] | TCTEX1D1 [58] | DNAH5 [35] | DNAL1 [35] | ||||
| LRRC6 | DNAI1, DNAI2 [37,38] | DNAH7 DNALI2 [37,38] | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabczak, H.; Osinka, A. Role of the Novel Hsp90 Co-Chaperones in Dynein Arms’ Preassembly. Int. J. Mol. Sci. 2019, 20, 6174. https://doi.org/10.3390/ijms20246174
Fabczak H, Osinka A. Role of the Novel Hsp90 Co-Chaperones in Dynein Arms’ Preassembly. International Journal of Molecular Sciences. 2019; 20(24):6174. https://doi.org/10.3390/ijms20246174
Chicago/Turabian StyleFabczak, Hanna, and Anna Osinka. 2019. "Role of the Novel Hsp90 Co-Chaperones in Dynein Arms’ Preassembly" International Journal of Molecular Sciences 20, no. 24: 6174. https://doi.org/10.3390/ijms20246174
APA StyleFabczak, H., & Osinka, A. (2019). Role of the Novel Hsp90 Co-Chaperones in Dynein Arms’ Preassembly. International Journal of Molecular Sciences, 20(24), 6174. https://doi.org/10.3390/ijms20246174