1. Introduction
Basement membranes (BM) are sheet-like, cell-adherent extracellular matrices (ECM) consisting of an enmeshed polymer of laminins and collagen IV (Col IV) that are bound to nidogens, agrin, and perlecan [
1]. They serve as both supportive cell substrata and solid-phase agonists, contributing tissue organization, stability, and differentiation. Col IV is essential for basement membrane (BM) stability [
2], and laminins are critical to the general scaffolding of BM. Col IV is assembled by extracellular oligomerization of triple-helical protomers and is covalently cross-linked by sulfilimine (S=N) bond formation among the C-terminal non-collageneous 1 (NC1) domains that reinforce the structural integrity of the Col IV networks [
3]. Ablation of both Col IV subunit α1 and α2 in mice causes lethality between E10.5 and E11.5 because of structural deficiencies in the BMs and eventual failure of the integrity of Reichert’s membrane [
2]. A dominant mutation of Col IV subunit chain α1 in mice results in BM defects that affect the eye, kidney, and other tissues [
4]. In humans, Col IV α1 mutation is also associated with abnormalities of the anterior chamber of the eye, vascular leukoencephalopathy, and hemorrhagic stroke [
5,
6,
7]. Col IV α2 mutations in mice and humans also cause diseases similar to those resulting from Col IV α1 mutations [
8,
9].
Peroxidasin (PXDN) is an ECM protein with peroxidase activity, which catalyzes formation of the sulfilimine bond between a methionine sulfur and hydroxylysine nitrogen using hypohalous acids in Col IV [
10]. Loss or decrease of PXDN severely disrupts organogenesis in
Drosophila,
C. elegans, and zebrafish by impairing basement integrity, leading to death at embryonic and larval stages (
Drosophila and C. elegans) or severely defective phenotypes, which consist of cardiac edema, decreased eye size, and gross trunk patterning defects (zebrafish) [
10,
11,
12]. The sulfilimine bond of Col IV is considered to contribute to reinforcing the mechanical strength of BM and maintaining tissue integrity. In mice and humans, much as in Col IV mutations, it can be postulated that PXDN mutations can cause BM-linked diseases, more specifically, Col IV-associated diseases, such as porencephaly-1, intracerebral hemorrhage, retinal arterial tortuosity, or anterior segment dysgenesis (ASD) [
4,
13]. Indeed, some homozygous mutations in
PXDN in humans cause severe inherited eye disorders, such as congenital cataracts, corneal opacity, and developmental glaucoma because of ASD; other recessive mutations in
PXDN show a broader phenotype, including ASD, sclerocornea, microphthalmia, hypotonia, and developmental delays [
14,
15].
Pxdn mutation-induced congenital eye diseases, such as ASD and microphthalmia, were also revealed in recessive mutant mice induced by
N-ethyl-
N-nitrosourea (ENU) [
16]. The mutant mice highly resemble the manifestations in patients with
PXDN mutations. However, these pathogenic phenotypes are milder than expected from the studies of lower animals, in which PXDN depletion causes embryonic and larval lethality or severely defective phenotypes. In addition, it has not been established yet how inactivation of the PXDN gene affects tissue genesis and organ development.
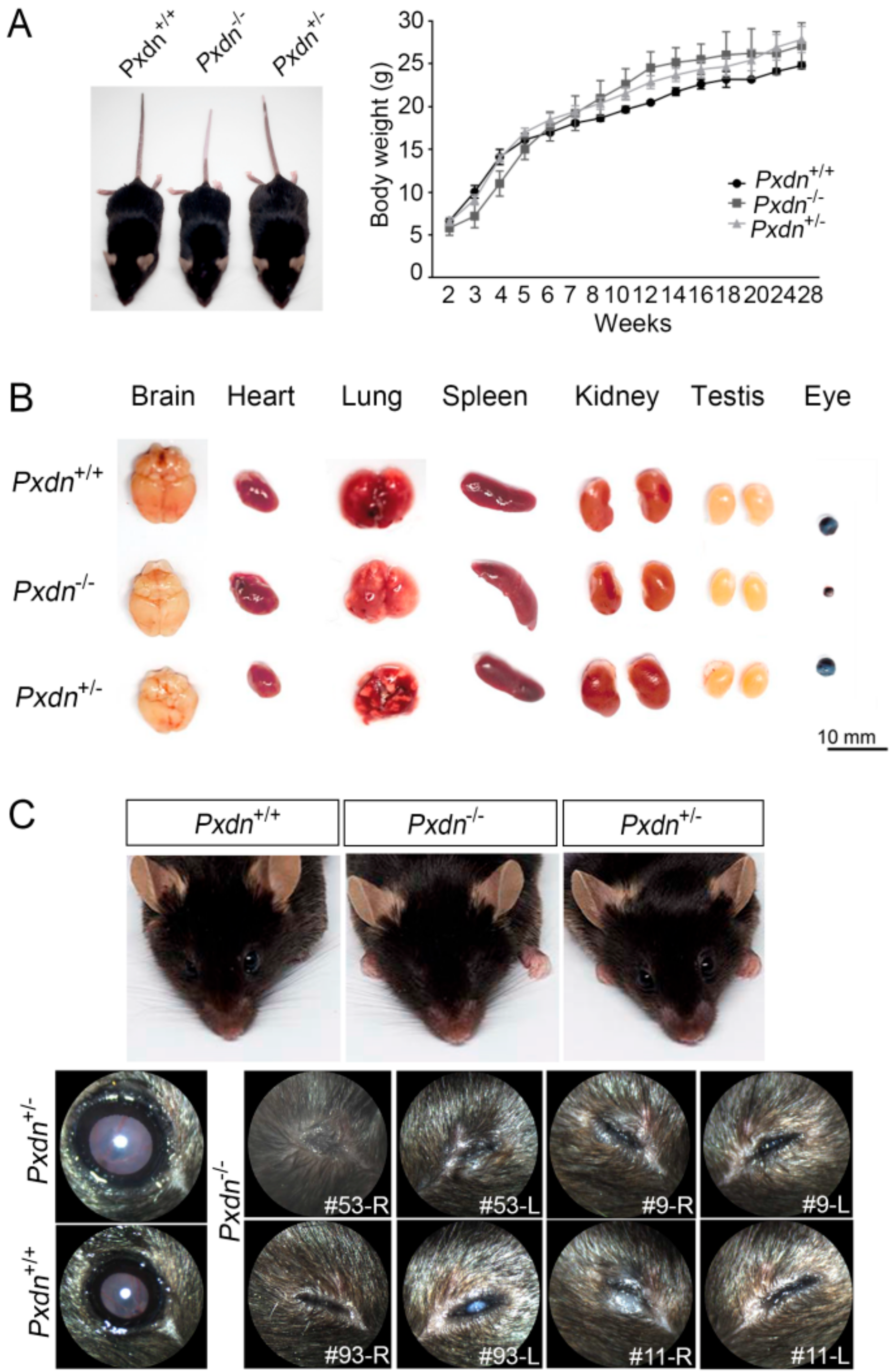
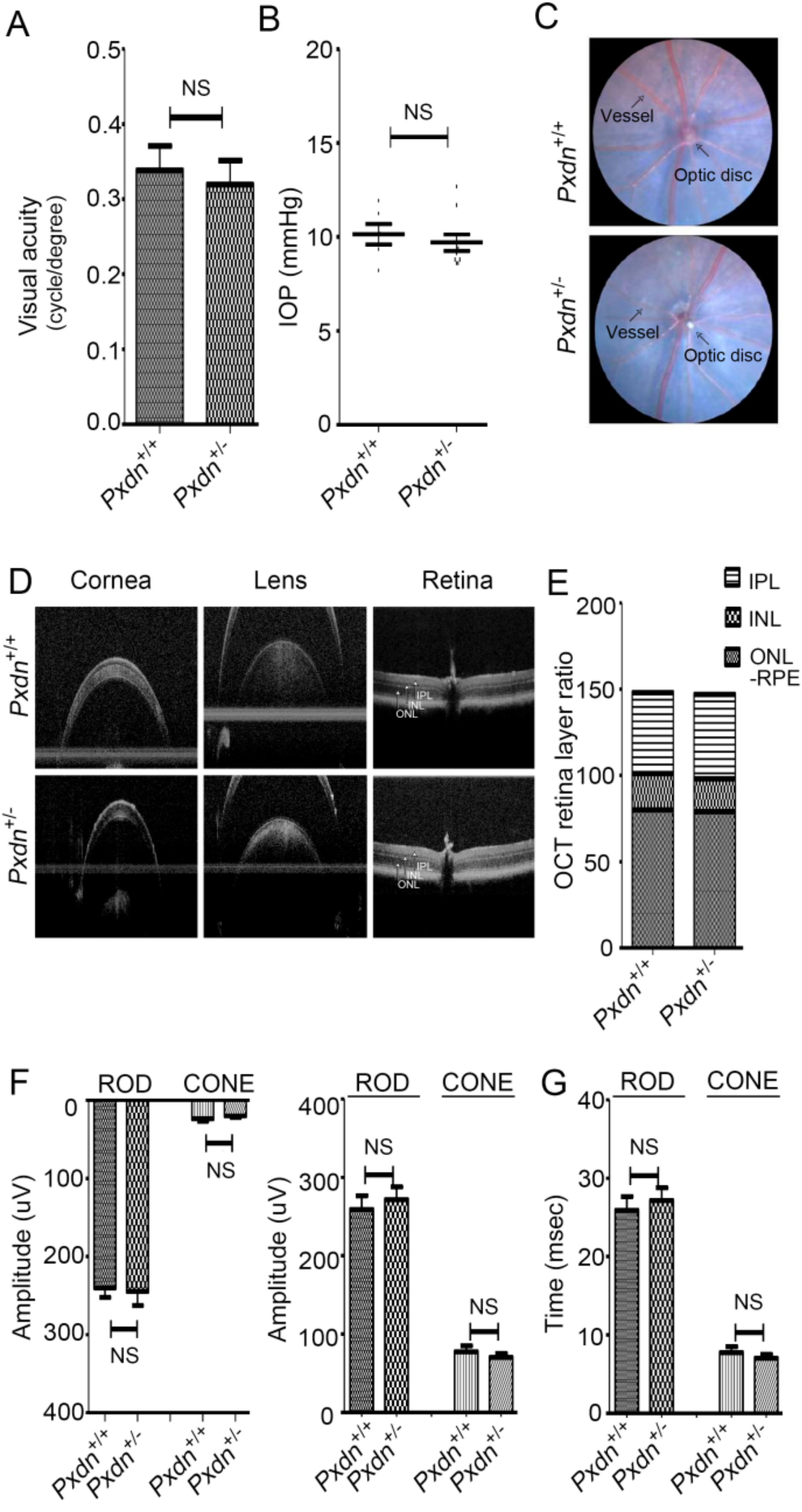
Here, we generated a knockout mouse model by deletion a sequence containing exon1 and its 5′ upstream sequences of the Pxdn gene using a CRISPR/Cas9-based genome editing system and analyzed the phenotypes of homozygous/heterozygous mice. We found that the homozygous mice had no eyes or extremely disorganized eye structures, whereas the heterozygous mice underwent development of normal eyes with proper visual functions. Grossly, no external morphological defect was detected in other organs. This study provides experimental evidence that PXDN is critical for eye development, especially in constructing precise eye structures, and that PXDN is haplosufficient for eye structure formation and normal visual function. This study also suggests that Pxdn knockout mice can be used as a novel mouse model for anophthalmia and severely malformed microphthalmia.
3. Discussion
Because PXDN is known to catalyze the formation of the sulfilimine crosslink that stabilizes the Col IV network for mammalian tissue genesis [
19], we suspected there would be embryonic lethality or severe impairment of development in
Pxdn-null mice. However, unlike our speculation, the
Pxdn KO mice did not show any apparent external morphological changes compared to those of the wild-type mice except for the abnormal ocular phenotypes, anophthalmia, and severely malformed microphthalmia. Previously, it had been reported that the ENU-induced
Pxdn mutation that has a premature stop codon (Cys1272X) leads to severe anterior segment dysgenesis and microphthalmia [
16]. In humans, the patients with several inherited mutations of the
PXDN gene also display eye disorders at various levels [
14,
15]. Such a phenotype shown by PXDN mutations in humans and mice was displayed in the
Pxdn KO mice. In the
Pxdn KO mice, deletion of exon1 and its 5′ upstream sequences of the
Pxdn gene resulted in complete gene inactivation by showing no mRNA/protein expression of PXDN in the tissues. On the other hand, the
Pxdn mutation with the premature stop codon (Cys1272X) was predicted to cause loss of function of peroxidase enzyme activity with the loss of the von-Willebrand factor type C (vWFC) domain [
16]. In their data, an immunofluorescence study showed that PXDN is expressed in the lens and the inner neuroblast layer of mutant eyes at E17.5 with an expression pattern similar to that of wild types. The peroxidase inhibitor phloroglucinol inhibits formation of sulfilimine crosslinks by PXDN at the cellular level [
10], and mutagenesis studies have also revealed that peroxidase activity is required for sulfilimine-bond formation by PXDN [
20,
21]. On the other hand, it cannot be excluded that ECM motifs can have their own function. Thus, it should be clarified in the future whether this point-mutated mouse expresses the truncated form of PXDN with loss of peroxidase activity and vWFC domain, and whether N-terminal ECM motifs can complement PXDN deficiency at some levels in mice. In this study, although
Pxdn-null mice showed severely defective eye phenotypes, the heterozygous mice with only a single intact allele did not show any defect in structure and visual function of eyes. Thus, we clearly showed that one allele of the
Pxdn gene is sufficient for eye structure formation and visual function.
Because a number of ophthalmological differences and sequence variants between C57BL/6J and C57BL/6N strains were identified from the comparative phenotypic and genomic sequence analysis [
22], it cannot be ruled out that the different genetic backgrounds for the C1272X mutants (C57BL/6J) and the KO mice (C57BL/6N) may contribute to the differences in phenotype. However, the comparison of the general visual functions of the two strains shows only reduced vision in C57BL/6N mice compared with C57BL/6J mice, not reflected relative to differences in lens opacities in both strains. Thus, severe ocular malformation by biallelic deletion of
Pxdn is not likely strain-dependent, but further studies are needed in future for clarification on this point.
Like the mutant mice with this premature stop codon (Cys1272X), the
Pxdn KO mice had a white spot at the ventral and/or dorsal region, and their tail color was white in two-thirds of the tip [
16]. The previous report noted that this phenotype is reminiscent of some mutations affecting migration of neural crest cells, which also affects eye development [
16,
23,
24]. Thus, it is interesting to figure out how
Pxdn gene function is correlated with neural crest cell migration.
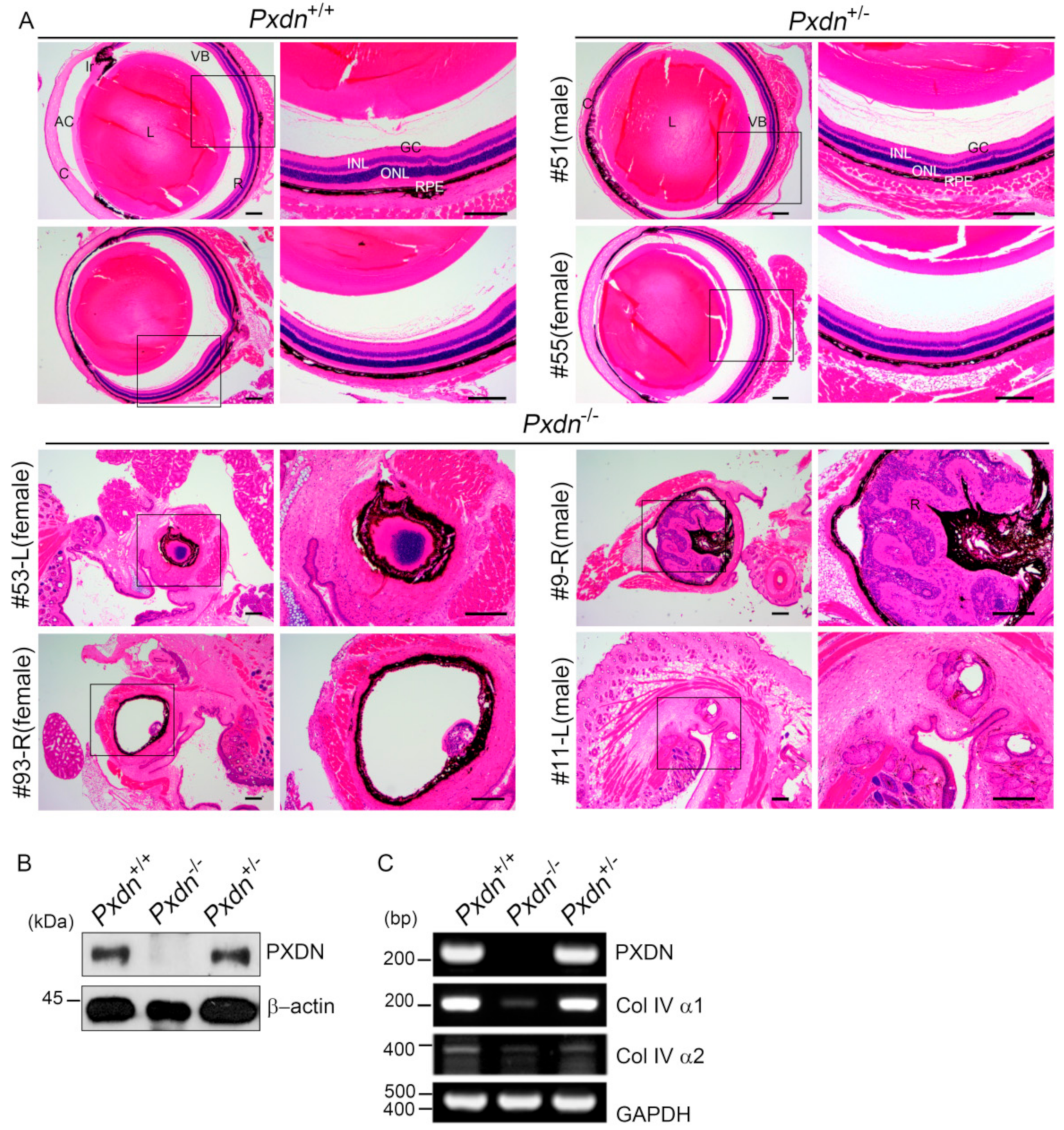
The lens capsule is a modified BM, which is important for structure, biomechanics, and maintenance of the lens cell phenotype [
25]. Some core structural molecules, including Col IV, laminin, and nidogen, provide protective and signaling roles for the mature lens. Col IV point mutants act as dominant negative molecules and affect synthesis and secretion of Col IV or the function of the Col IV network, which leads to defects of the kidneys, esophagus, aorta, and eyes [
4]. Therefore, BM defects can lead to damage to cell and tissues, developmental defects, and diseases [
19]. The decrease of Col IV expression level in the eye tissue of the
Pxdn KO mice compared with that of the WT mice may be because of ocular developmental defects including missing lenses, as Col IV is highly expressed in lens capsules [
26]. From this study, PXDN deficiency is presumed to be associated with developmental disability in eyes, as they lack the stability of the Col IV network because of the deficiency of sulfilimine crosslinks. The defect in eye phenotype of Col IV mutant mice, such as iris/corneal adhesion, is similar to that of the
Pxdn mutant mice [
16], and the
Pxdn KO mice showed highly disorganized eye structure or no eyeballs. Therefore, the sulfilimine crosslinks in Col IV may be very important in the development of eye tissue, which requires a complex, precise structure for precise functioning of the eyes. In addition, this fact suggests the importance of the BM integrity generated by PXDN, which is responsible for Col IV network stability. However, it is notable that
Pxdn gene inactivation by deletion of exon1 and its 5′ upstream sequences leads to no apparent developmental defect in kidney, esophagus, or aorta, which is different from the defects of Col IV mutant mice [
4]. A recent study using
Pxdn KO mice generated by exon 9 deletion also showed no alteration of BM structure in the renal glomeruli or tubules, but reported a finding that reduced Col IV sulfilimine crosslinks reduced renal tubular BM stiffness [
27]. This finding corresponds with our finding suggesting the importance of BM integrity driven by PXDN.
Considering that the sulfilimine bond is presumed to provide the mechanical strength for the Col IV network, it is still questionable as to why it is important in the development of the eyes. Anophthalmia, the most severe phenotype, is associated with mutations of the
Rax, Sox2, Lhx2, BMP-4, and
BMP7 genes [
28]. Complete inactivation of these genes also displays embryonic lethality because of broad impacts on development.
Pax-6 homozygous mutants, the classical model for anophthalmia in the mouse, do not develop eyes, but die perinatally because of other developmental defects, including those of the central nervous system [
29,
30]. In the future, it will be interesting to address why the contribution of PXDN in tissue genesis of mice is markedly significant only for the eyes, but not for other organs, such as the kidneys and vessels.
From this study, we can suggest that the Pxdn gene is haploid sufficient for normal eye development and visual function, providing evidence that PXDN is essential for eyeball development. It will be also interesting to investigate in the future whether PXDN is correlated with the pathogenesis of glaucoma and cataract in adult eyes after normal eye development.
4. Materials and Methods
4.1. CRISPR/Cas9 for Pxdn Gene Knockout
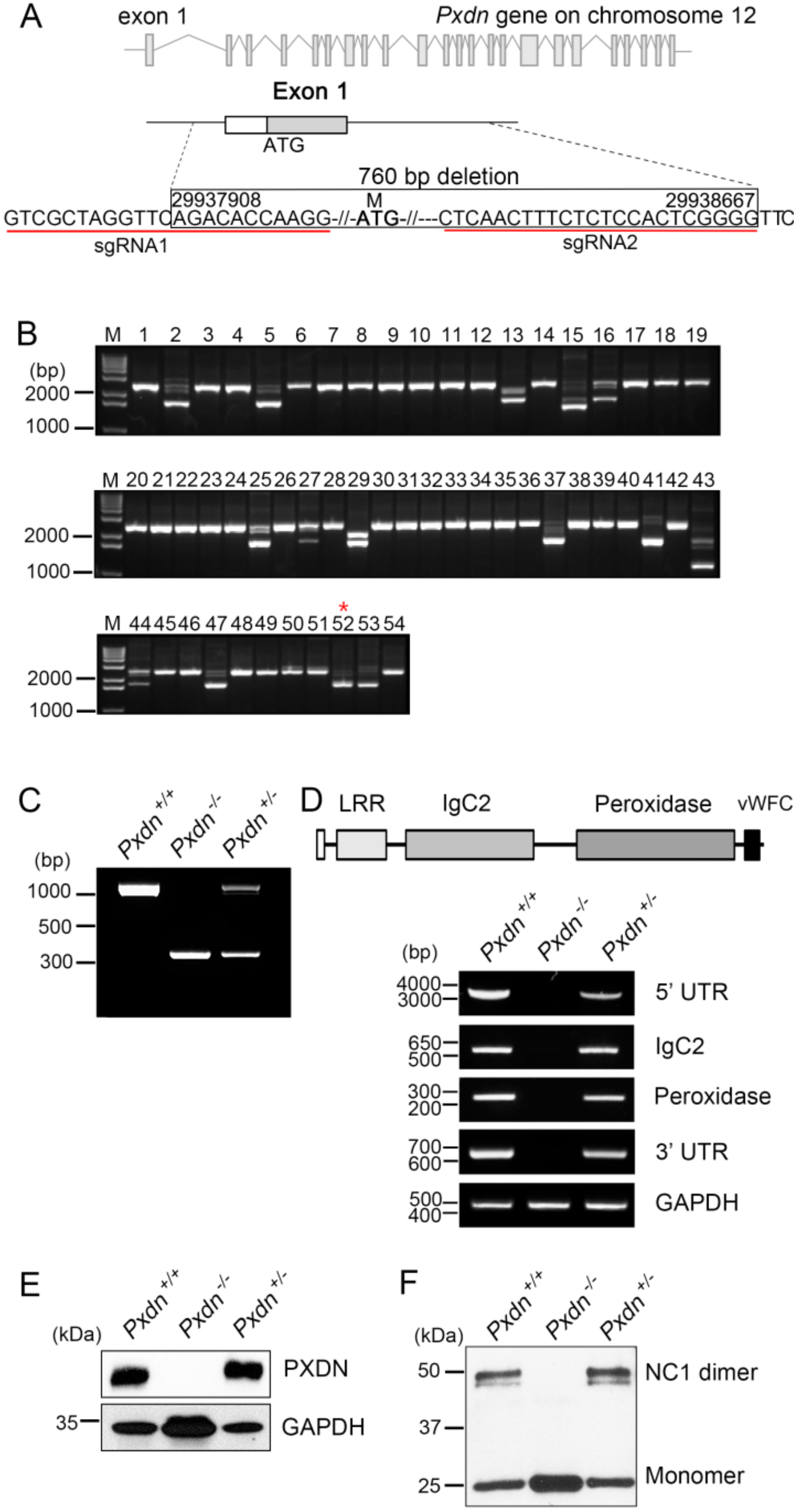
We purchased SpCas9 protein and sgRNAs to generate Pxdn knockout mice from Macrogen, Inc. (Seoul, Korea). To knock out the Pxdn gene (NC_000078.6), we designed two sgRNAs (1 (5′ UTR gR3), GTCGCTAGGTTCAGACACCAAGG; 2 (intron1-2-gR2), CTCAACTTTCTCTCCACTCGGGG) to target the exon1 and its 5′ upstream sequences of the Pxdn gene and validated them using the T7E1 in vitro cleavage reaction of template DNAs, which were amplified by PCR (F1:5′-TGTGGGAACAGTGGTGGGCA-3′ and R1: 5′-GTGAACAGCAGCGGAGGAG-3′, F2: 5′-CAGGGGTACTGGTTTGGAGA-3′ and R2: 5′-CCAAGCAAAGAAAAAGCTCCT-3′). Briefly, the amplified template DNA was incubated for 90 min at 37 °C with Cas9 protein (20 nM) and sgRNA (40 nM) in 1× NEB 3 buffer. Reactions were stopped with 6× stop solution containing 30% glycerol, 1.2% SDS, and 100 mM EDTA. Cleavage activity was confirmed by electrophoresis of the reaction mixture.
4.2. Generation of Pxdn Gene Knockout Mice
Pxdn knockout mice were generated by Macrogen, Inc., and were interbred and maintained in a pathogen-free condition at Macrogen, Inc. (Seoul, Korea). All animal experiments were performed in accordance with the Korean Food and Drug Administration (KFDA) guidelines. Protocols were reviewed and approved by the Institutional Animal Care and Use Committees (IACUC) of Macrogen, Inc. (MS-2016-01(5 October 2016)). All manipulations were conducted with the Institutional Animal Care and Use Committee approval. Briefly, pregnant mare serum gonadotropin (PMSG) and human chorionic gonadotropin (hCG) were injected into C57BL/6N female mice. After 48 h, these female mice were mated with C57BL/6N stud male mice. Next day, virginal plug-checked female mice were sacrificed and fertilized embryos were harvested. The mixture of sgRNA and SpCas9 protein was microinjected into one-cell embryos, and microinjected embryos were incubated at 37 °C for 1–2 h. Then, 14 to 16 injected one-cell-stage embryos were transplanted into oviducts of pseudopregnant recipient mice (ICR). After F0 mice were born, genotyping was done by direct PCR and sequencing methods using tail-cut samples (SF3 primer, 5′-TGGCAGAACTGGGTAACTCC-3′; and SR3 primer, 5′-CAATCTCCCCCAGAGGAAGT-3′). Among of the founders with altered sequences, we selected F0 mice with deletion of exon1 and its 5′ upstream sequences of Pxdn.
4.3. Genotyping
Genomic DNA was isolated from the lung tissues of each mouse using an MGmed tissue kit (MGmed, Seoul, Korea). Genotyping was done by PCR amplification using the following primers: SF1, 5′-AAGCAGAATCAGGGATCCAA-3′; and SR2, 5′-AGCCGATGGGAGTACACTTG-3′. The PCR products were visualized by agarose gel electrophoresis; a 1096 bp fragment was amplified in the wild-type mice, both 1096 bp and 336 bp fragments in the heterozygous mice, and a 336 bp fragment in Pxdn-null mice.
4.4. RT-PCR Analysis
Expression of PXDN in the tissues was confirmed by RT-PCR analysis. Total RNA was extracted from the tissues by Trizol reagent (Invitrogen, Carlsbad, CA, USA). We performed cDNA synthesis using the SuperScript synthesis system for RT-PCR (Enzynomics, Daejeon, Korea) according to the manufacturer’s instructions. The primer sets used for PXDN were as follows: for 5′ UTR-exon 19, forward, 5′-GACCGGAGGGCTCAGTTG-3′ and reverse 5′-ACACGGGTGATGTTGTCTGA-3′; for exon 10–14 (IgC2 3-4 domain), forward 5′-CGCGCGGCAGCCATATGGCCCTTCTCAGTTCACT-3′ and reverse 5′-GTTAGCAGCCGGATCCTCAATTCACACTGAGTACCATGCT-3′; for exon 17–19 (peroxidase domain), forward 5′-GGCTGTATGGCTCGACTCTC-3′and reverse 5′-ACACGGGTGATGTTGTCTGA-3′; for 3′ UTR, forward 5′-GGCTAGGGAGGAAGACCTCA-3′ and reverse 5′-CCTGGGCAATGAGGCTGTAA-3′; for GAPDH, forward 5′-CATGACCACAGTCCATGCCATCACT-3′ and reverse 5′-TGAGGTCCACCACCCTGTTGCTGTA-3′. The following primers were used to measure the mRNA expression level of Col IV: forward 5′-GCCAAGTGTGCATGAGAAGA-3′ and reverse 5′-AGCGGGGTGTGTTAGTTACG-3′ for Col IV α1, forward 5′-CCGATTCCACGAGCCCCTT-3′ and reverse 5′-CTCCTTTCTCCGGGTAGCAC-3′ for Col IV α2. PCR amplification for PXDN mRNA expression was done under the following conditions: 95 °C for 1 min, followed by 28–33 cycles of 1 min at 95 °C, 1 min at 56–64 °C, and 1–4 min at 72 °C. The PCR product was loaded in 1–2% agarose gel mixed with SYBR safe dye (Invitrogen).
4.5. Preparation of Anti-Mouse PXDN Antibody
The recombinant peroxidasin IgC2 (3–4) subdomain (amino acids 428–609) of mouse PXDN was prepared for antibody production. A 546 bp DNA fragment encoding amino acids spanning from Ala (428) to Asn (609) of mouse PXDN was amplified by PCR using mouse lung cDNA,
Pfu polymerase, and primers (forward: 5′-CGCGCGGCAGCCATATGGCCCTTCTCAGTTCACT-3′, reverse: 5′-GTTAGCAGCCGGATCCTCAATTCACACTGAGTACCATGCT-3′) and subcloned between the
NdeI and
BamHI sites of pET-15b (Novagen, Madison, WI, USA). Then, the recombinant protein was expressed in
E. coli BL21 (DE3) and purified from the cell pellet as described previously [
31]. Polyclonal antibody was raised in rabbits against the purified recombinant protein (Abfrontier, Seoul, Korea).
4.6. Western Blot Analysis
For protein preparation, the tissues were dissected and lysed using lysis buffer (50 mM Tris (pH 8.0), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 mM Na3VO4, 50 mM NaF, 1 mM EDTA, 1 mM EGTA, 2 mM PMSF, 1 μg/mL pepstatin, and protease inhibitor mix (Roche, Basel, Switzerland)). The lysate was centrifuged at 14,000 rpm for 30 min, and then the supernatant was subjected to 8% SDS-PAGE. Separated proteins were transferred to a nitrocellulose membrane, and the membrane was blocked with 5% skim milk. Blots were incubated with polyclonal anti-mouse PXDN antibodies raised against recombinant IgC2 (3–4) subdomain, and then with horseradish peroxidase (HRP)-conjugated secondary antibodies. GAPDH or β-actin was used for a loading control. We visualized the immunoreactive bands using a chemiluminescent substrate (ECL kit) (Amersham, Amersham, United Kingdom).
4.7. Detection of NC1 Sulfilimine Crosslink
We assessed the level of NC1 domain sulfilimine crosslinking in Col IV according to a procedure reported previously [
32]. Briefly, the lung tissues were lysed in hypotonic lysis buffer (10 mM CaCl
2, 50 mM HEPES, pH 7.4, 0.1 mM benzamininde hydrochloride, 25 mM 6-aminocaproic acid, and 1 mM PMSF). The tissue lysate was digested with 0.5 mg/mL collagenase (Gibco) at 37 °C for 18 h and then subjected to SDS-PAGE under reducing conditions followed by western blotting. Crosslinked dimeric and un-crosslinked monomeric NC1 domains were detected with anti-Col IV α2 antibody (ChondrexInc, Redmond, WA, USA).
4.8. Hematoxylin and Eosin Staining
For the histological examination, the eye specimen was fixed with 4% Davidson’s solution for 24 h with gentle shaking. Fixed samples were paraffin-embedded and cut into 5 μm sections using a microtome (RM2335, Leica, Wetzlar, Germany). Hematoxylin and eosin staining (H&E) was done after a day of sectioning. For H&E staining, paraffin sections were deparaffinized and then hydrated in a descending grade of ethanol. Next, we stained sections with 0.1% Mayer’s hematoxylin for 10 min and 0.5% eosin in 95% EtOH. After staining with hematoxylin and eosin, the washing steps were immediately and sequentially done as follows: dipped in distilled water until eosin stopped streaking, dipped in 50% EtOH 10 times, dipped in 70% EtOH 10 times, and incubated in 95% EtOH for 30 s and 100% EtOH for 1 min. Then, samples were covered with mount solution (6769007, Thermo Scientific, Waltham, MA, USA) and examined under the light microscope (BX43, OLYMPUS, Shinjuku, Japan).
4.9. Optokinetic Nystagmus (OKN)
All mice for visual function tests were in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC: 2018-0145) of Yonsei University. All animals were handled according to the principles of the Association for Research in Vision and Ophthalmology statement. We assessed spatial frequency thresholds (i.e., visual acuity) by optokinetic nystagmus (OKN) using a virtual optokinetic system (OptoMotry, Cerebral Mechanics, Medicine Hat, Alberta, Canada). A video camera located on the ceiling of the device recorded the images and sent them to the connected computer. The clockwise movement was to track the movement of the left eye, and the counterclockwise movement tracked the reaction of the right eye of the mouse. The experimenter judged whether the head and body of the mouse were tracking according to the direction in which the drift grid was rotating. If the presence of the tracking was unclear or absent, the retry was resumed, ignoring large repositioning and grooming movements. The maximum spatial frequency capable of driving head tracking was determined.
4.10. Tonometry
We anesthetized mice with an intraperitoneal injection of xylazine (10 mg/kg; Rompun, Bayer Animal Health, Leverkusen, Germany) and zolazepam and tiletamine (30 mg/kg; Zoletil 50, Vibrac, Carros, France). Intraocular pressure (IOP) was measured using a rebound tonometer (Icare TONOLAB tonometer, Colonial Medical Supply, Franconia, NH, USA). IOP measurements were taken according to the manufacturer’s instructions. One trial result was given after six consecutive measurements, and the mean of consecutive trials was used for analyses.
4.11. Optical Coherence Tomography (OCT)
We anesthetized mice with an intraperitoneal injection of xylazine (10 mg/kg; Rompun, Bayer Animal Health) and zolazepam and tiletamine (30 mg/kg; Zoletil 50, Vibrac, Carros, France). The pupils were dilated with 0.5% tropicamide and 0.5% phenylephrine mixed eyedrop (Mydrin-P, Santen Pharmaceutical Co, Ltd., Osaka, Japan). Optical coherence tomography (OCT) scans were taken using the Eyemera OCT (IISCIENCE, Busan, Korea). The cornea was lubricated with hypromellose 2.5% (Goniovisc, Hub Pharmaceuticals LCC, Roncho Cucamonga, CA, USA). After placing the mouse in front of the OCT lens and focusing on the retina, fundus photography and OCT scan were performed. Retinal thickness was measured using InSight-Animal OCT Segmentation Software (Phoenix Research Labs, Pleasonton, CA, USA). Each scan for cornea, lens, and retina centered on the optic nerve was obtained.
4.12. Electroretinography (ERG)
We performed ERG analysis using Micron Ganzfeld ERG (Phoenix Research Labs, Pleasanton, CA, USA). Mice were dark-adapted at least 12 h before the experiment for scotopic testing (rod-cell response). After anesthesia, the pupil was dilated as previously described. Once the pupil was fully dilated, we applied hypromellose 2.5% (Goniovisc) and inserted the electrodes. ERG was recorded as Ganzfeld ERG according to the standard protocol of the manual instruments. Scotopic ERG was obtained with increasing flash intensity in the range of −1.7 log cd/s/m2 to 1.9 log cd/s/m2. Mice were light-adapted for 15 min prior to the cone-cell response experiment. Photopic ERG was done with a flash intensity in the range of −0.5 log cd/s/m2 to 4.1 log cd/s/m2. A total of 10 responses to light stimuli were averaged. The implicit times of a-wave (as a measure of photoreceptor function), b-wave (as a measure of bipolar cell function), amplitude, and rod- and cone-cell response were determined.
4.13. Data and Statistical Analysis
Data were presented as means ± SEM of representative experiments. All the in vivo visual experiments were performed at least six times. Student’s t-test was used to calculate statistical difference between the two groups.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}



