Unexpected Racemization in the Course of the Acetalization of (+)-(S)-5-Methyl-Wieland–Miescher Ketone with 1,2-Ethanediol and TsOH under Classical Experimental Conditions

 , and
, and
Abstract
:
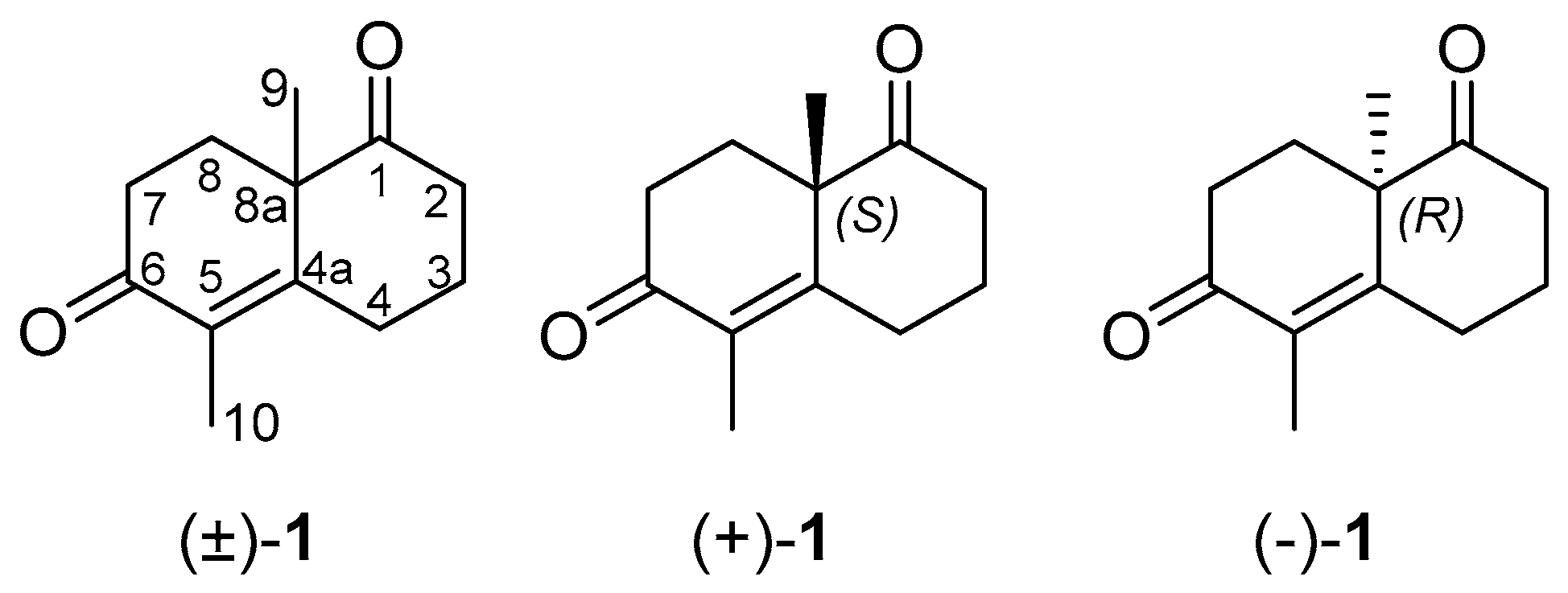
1. Introduction
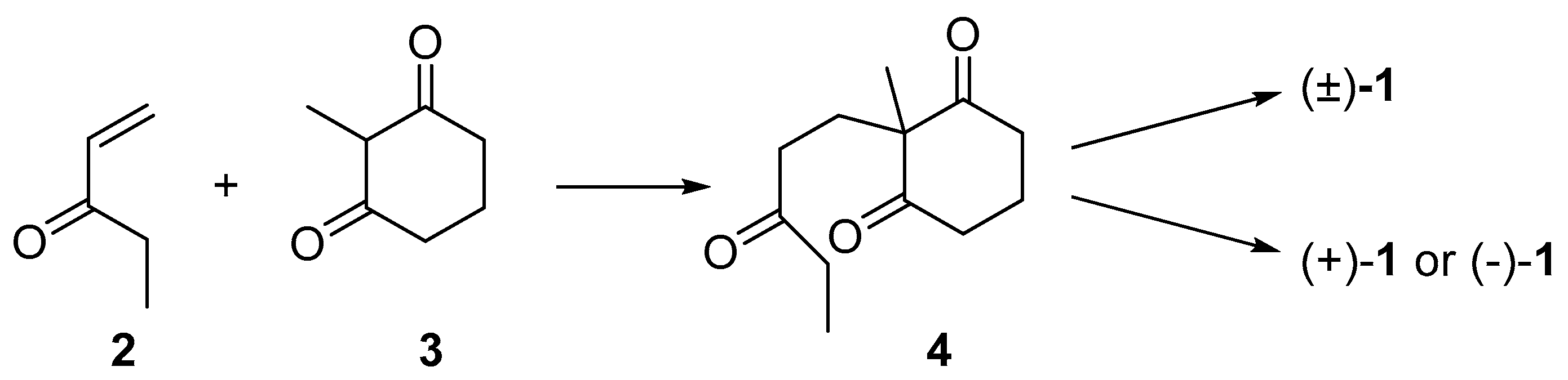
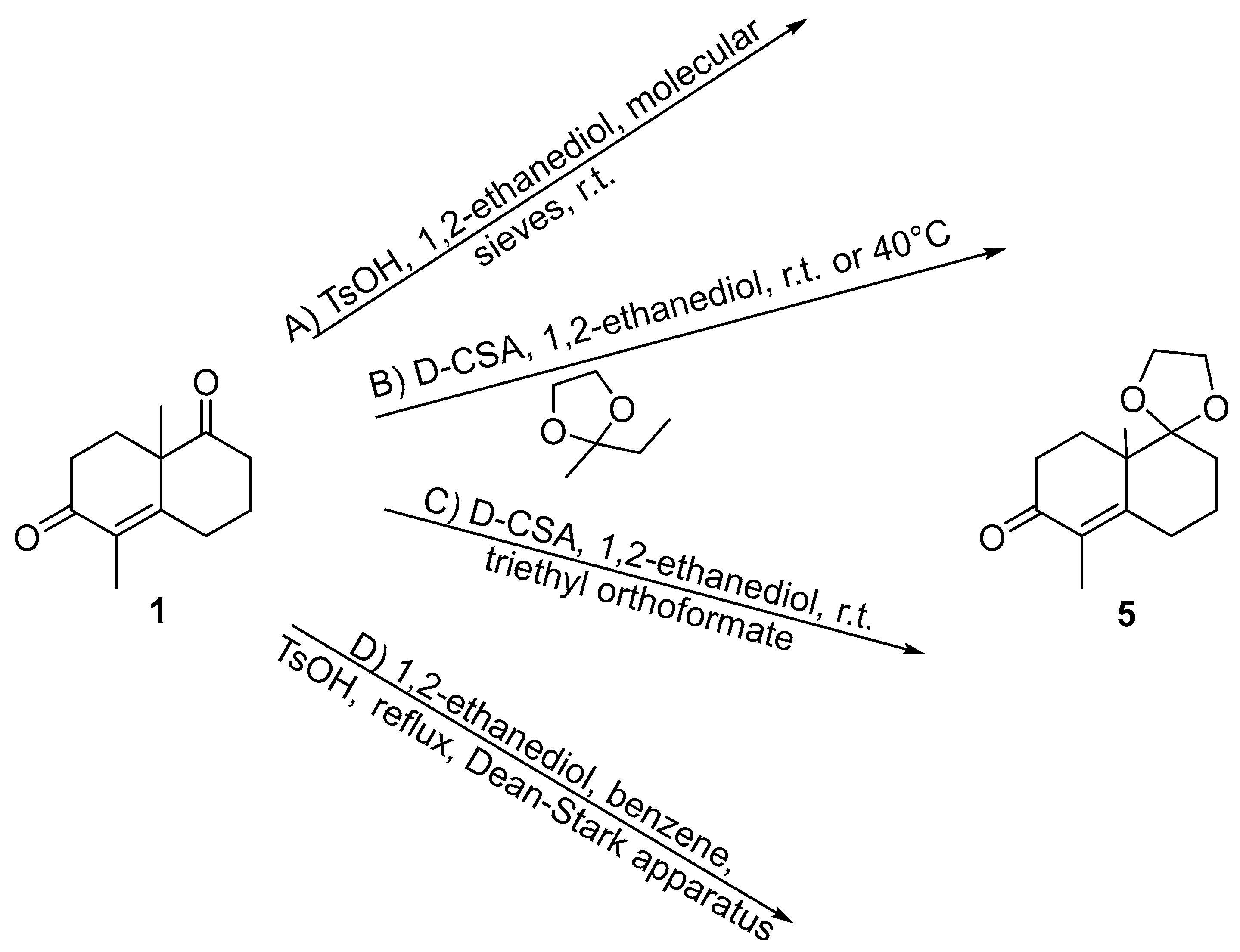
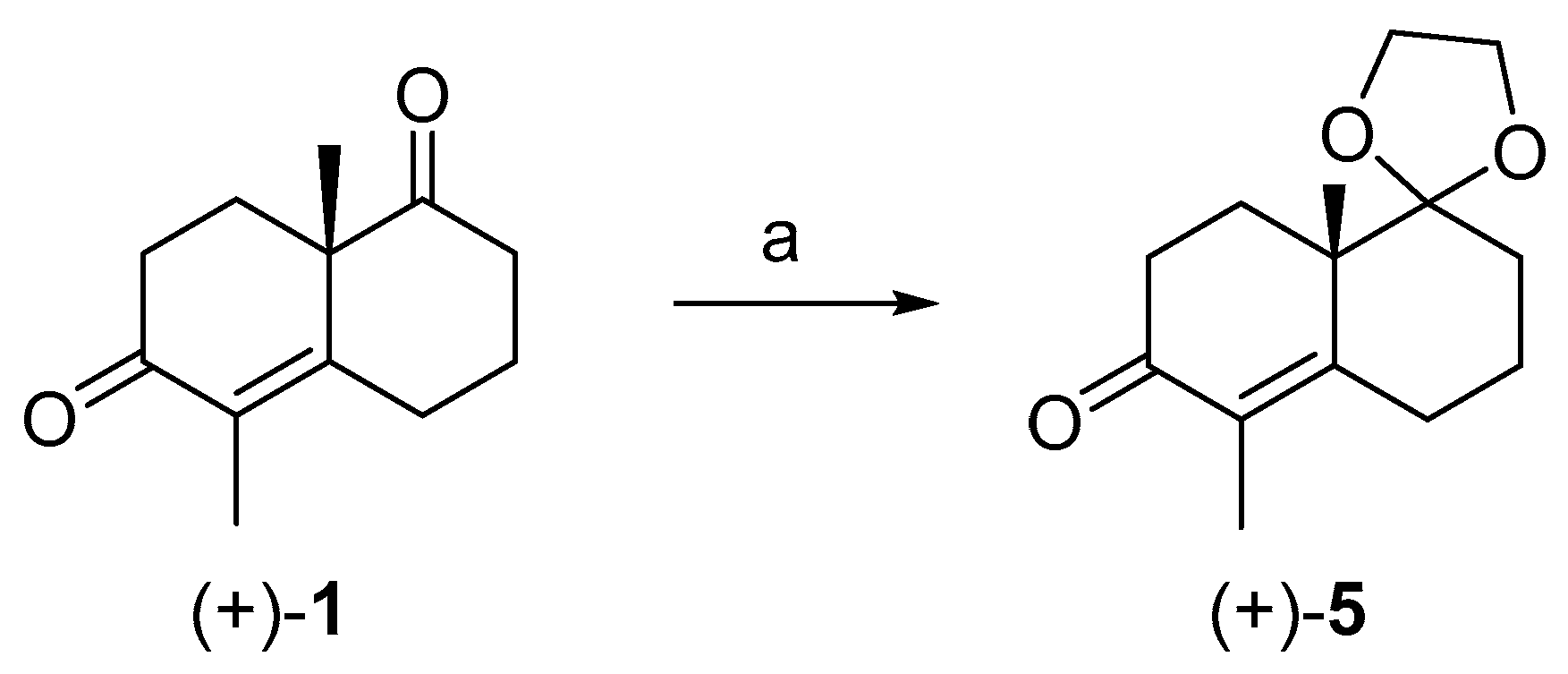
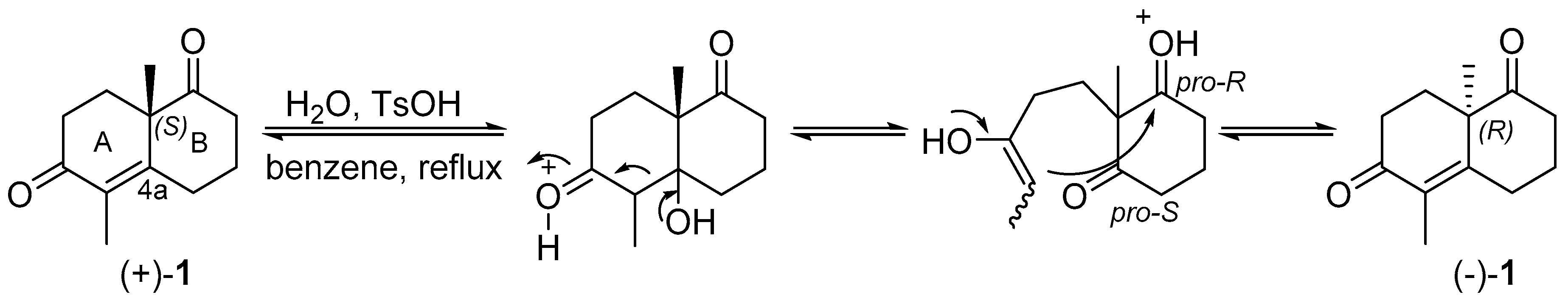
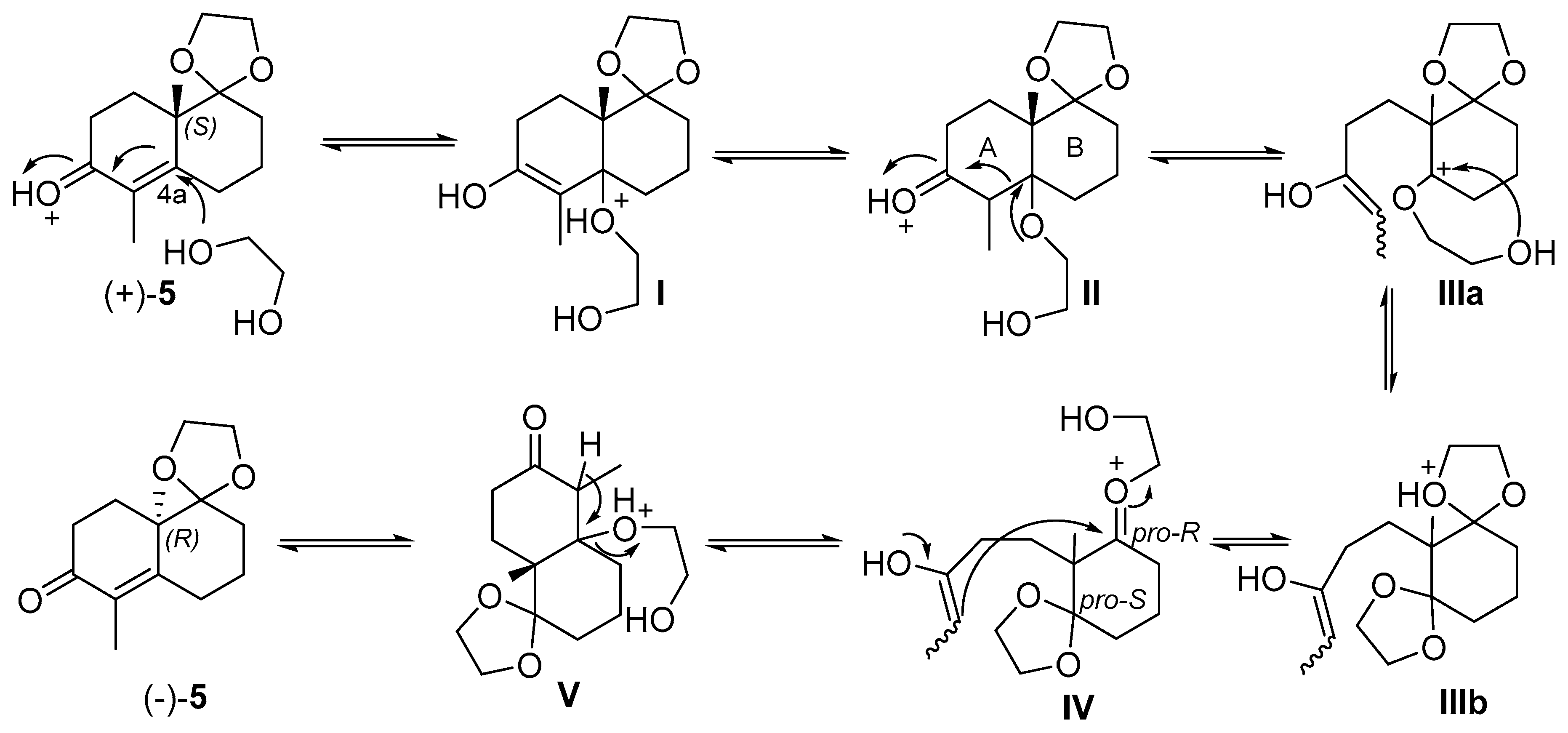
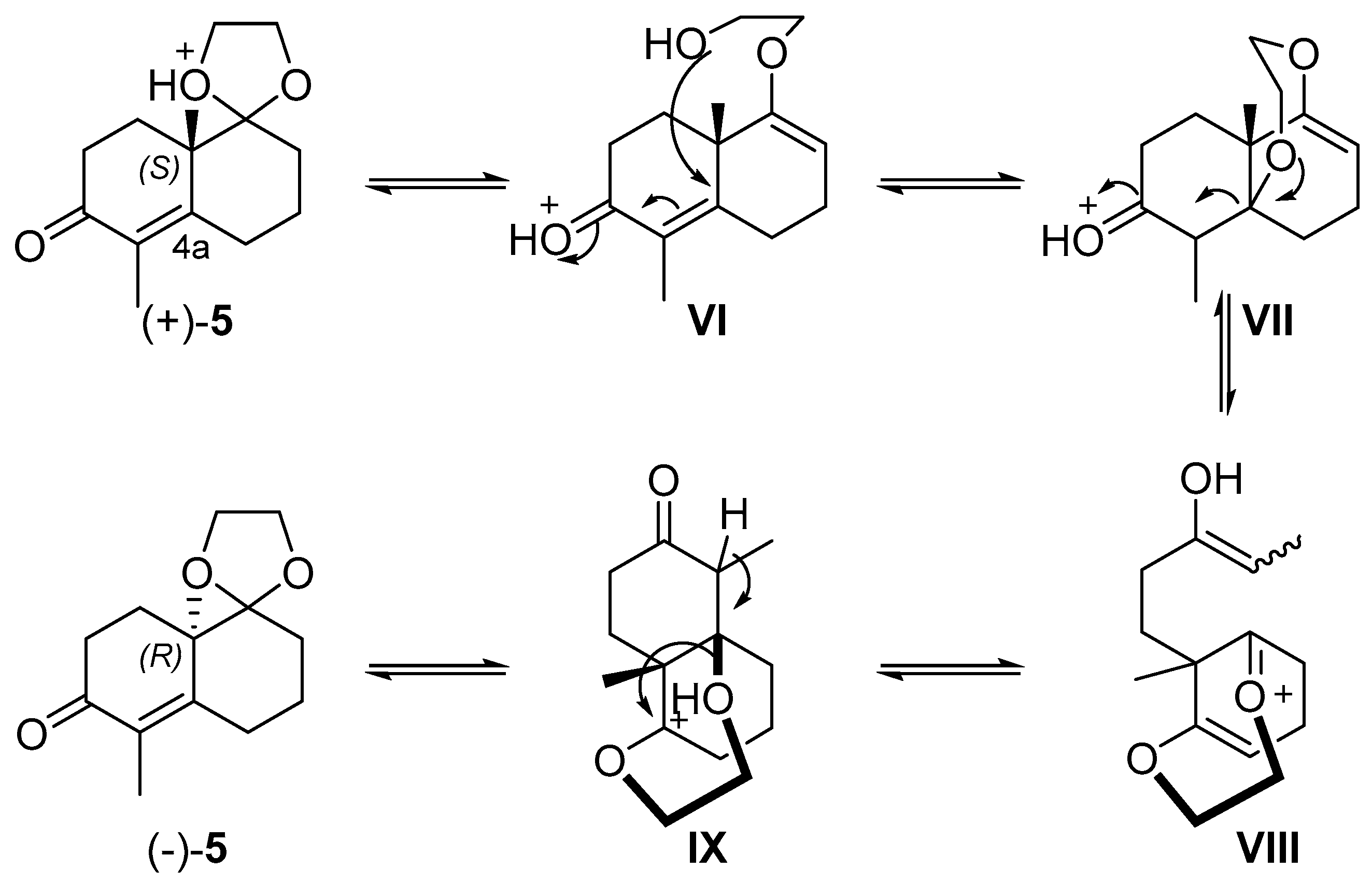
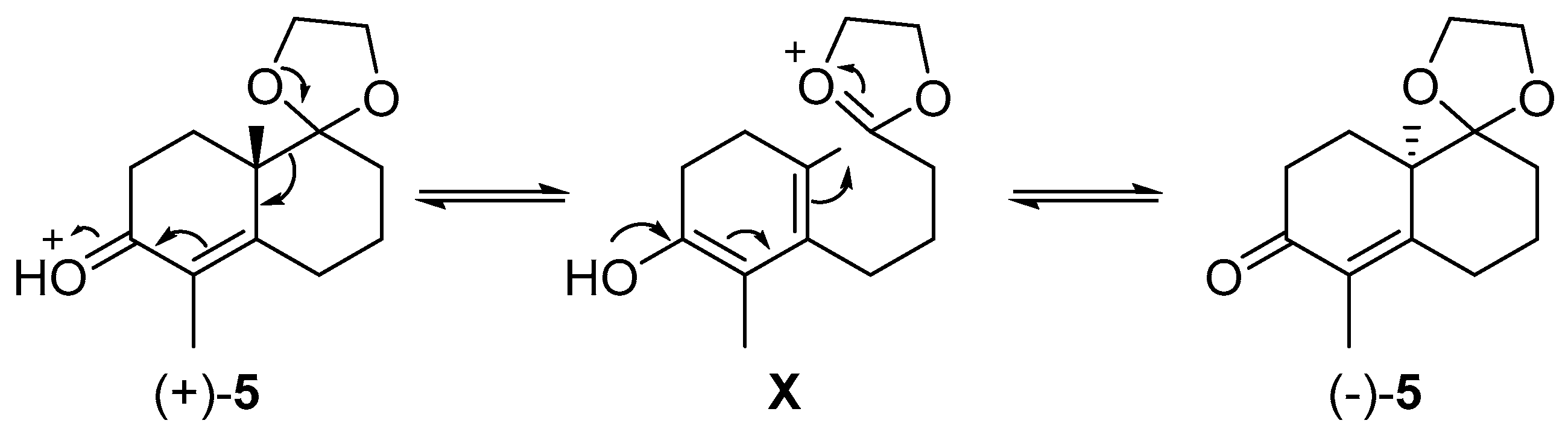
2. Results and Discussion
3. Materials and Methods
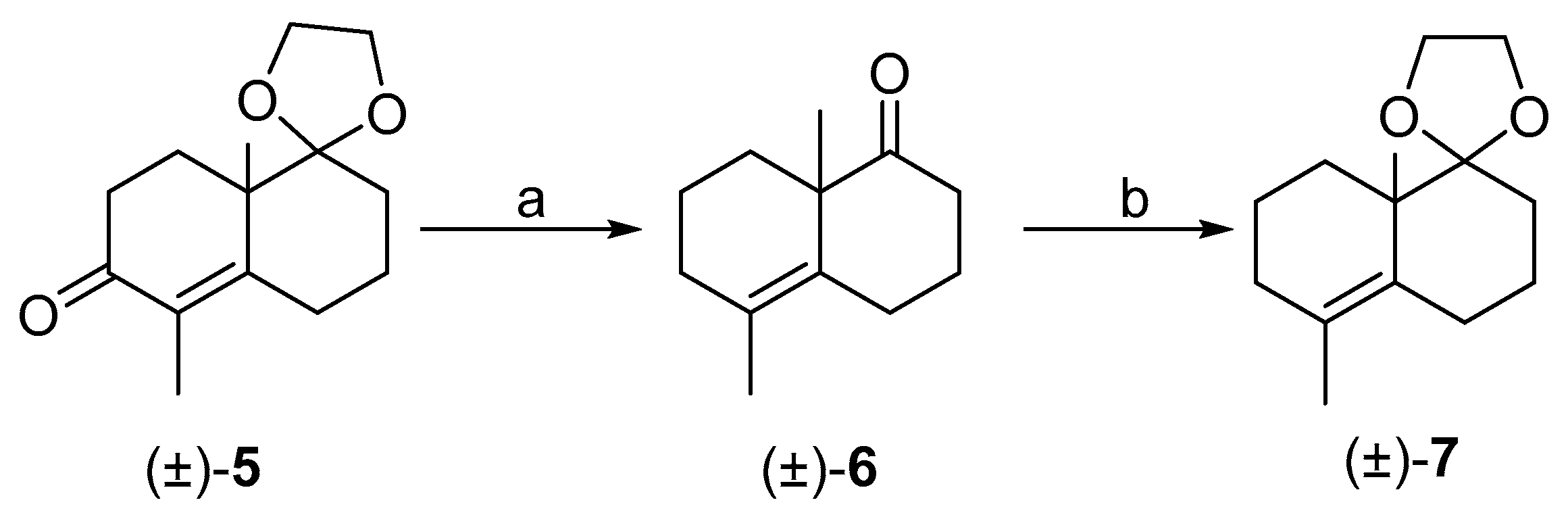
3.1. Synthesis of (±)-1
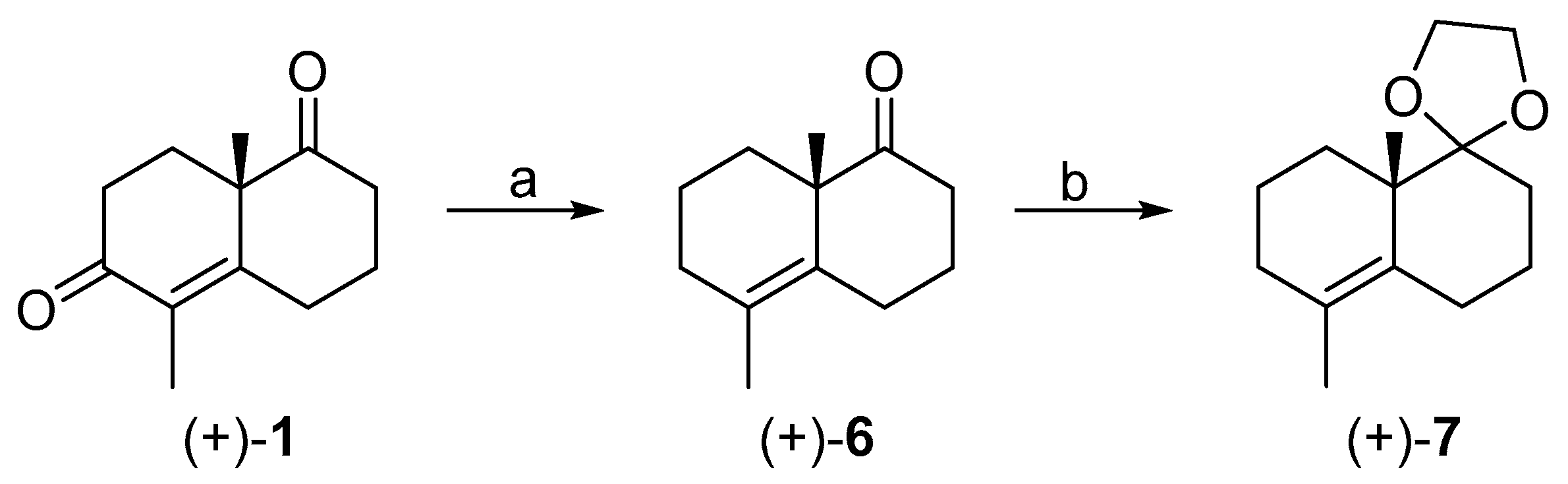
3.2. Synthesis of (+)-1
3.3. Synthesis of (±)-5 in Refluxing Benzene
3.4. Synthesis of (+)-5 at Rt
3.5. Acetalization of (+)-1 under Classical Condition in Refluxing Benzene
3.6. Synthesis of (±)-6
3.7. Synthesis of (+)-6
3.8. Synthesis of (±)-7
3.9. Synthesis of (+)-7
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kitahara, Y.; Yoshikoshi, A.; Oida, S. Total synthesis of dolabradiene. Tetrahedron Lett. 1964, 5, 1763–1770. [Google Scholar] [CrossRef]
- Trost, B.M.; Nishimura, Y.; Yamamoto, K.; McElvain, S.S. A total synthesis of aphidicolin. J. Am. Chem. Soc. 1979, 101, 1328–1330. [Google Scholar] [CrossRef]
- McMurry, J.E.; Andrus, A.; Ksander, G.M.; Musser, J.H.; Johnson, M.A. Stereospecific total synthesis of aphidicolin. J. Am. Chem. Soc. 1979, 101, 1330–1332. [Google Scholar] [CrossRef]
- Takahashi, S.; Kusumi, T.; Kakisawa, H. Total synthesis of (±)-annonene, a trans-clerodane diterpene. Chem. Lett. 1979, 8, 515–518. [Google Scholar] [CrossRef]
- Coisne, J.-M.; Pecher, J. A terpene synthesis intermediate: (±)-5-ethenyl-1,1,4aβ-trimethyl-1,2,3,4,4a,7,8,8aα-octahydronaphthalene. Bull. Soc. Chim. Belg. 1980, 89, 551–557. [Google Scholar] [CrossRef]
- McMurry, J.E.; Andrus, A.; Ksander, G.M.; Musser, J.H.; Johnson, M.A. Total synthesis of aphidicolin. Tetrahedron 1981, 37, 319–327. [Google Scholar] [CrossRef]
- Kende, A.S.; Roth, B. Stereospecific total synthesis of ajugarin-IV. Tetrahedron Lett. 1982, 23, 1751–1754. [Google Scholar] [CrossRef]
- Garver, L.C.; van Tamelen, E.E. Total synthesis of (±)-triptonide and (±)-triptolide. J. Am. Chem. Soc. 1982, 104, 867–869. [Google Scholar] [CrossRef]
- Marini Bettolo, R.; Tagliatesta, P.; Lupi, A.; Bravetti, D. A stereoselective total synthesis of (±)-maritimol, (±)-2-deoxystemodinone, (±)-stemodinone, and (±)-stemodin. Helv. Chim. Acta 1983, 66, 760–770. [Google Scholar] [CrossRef]
- Marini Bettolo, R.; Tagliatesta, P.; Lupi, A.; Bravetti, D. A Total synthesis of aphidicolin: Stereospecific synthesis of (±)-3α,18-dihydroxy-17-noraphidicolan-16-one. Helv. Chim. Acta 1983, 66, 1922–1928. [Google Scholar] [CrossRef]
- Liu, H.J.; Wynn, H. Thiol esters in organic synthesis. XIV. The total synthesis of racemic α-costal. Can. J. Chem. 1986, 64, 658–666. [Google Scholar] [CrossRef]
- Ziegler, F.E.; Hwang, K.J.; Kadow, J.F.; Klein, S.I.; Pati, U.K.; Wang, T.F. Practical routes to two functionalized decalones for the synthesis of quassinoids. J. Org. Chem. 1986, 51, 4573–4579. [Google Scholar] [CrossRef]
- Murae, T.; Sasaki, M.; Konosu, T.; Matsua, H.; Takahashi, T. Synthesis of bruceantin skeleton. Tetrahedron Lett. 1986, 27, 3411–3414. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Peña Matheud, C.A.; Hurtado, H.E.; Díaz, M.G. Synthesis of (±)-1β-methoxy-9β,7β-epoxy-5α-methyl-trans-decalin. Heterocycles 1986, 24, 2155–2163. [Google Scholar] [CrossRef]
- Lupi, A.; Patamia, M.; Marini Bettolo, R. A Total synthesis of (±)-aphidicolin: Regio and stereoselective conversion of 3α,18-Di-O-benzyl-17-nor-14-aphidicolen-16-one into (±)-aphidicolin. Helv. Chim. Acta 1988, 71, 872–875. [Google Scholar] [CrossRef]
- Inayama, S.; Shimizu, N.; Ohkura, T.; Akita, H.; Oishi, T.; Itaka, Y. Microbiologically modified chiral synthon. I. 3,8-dioxo-4-methoxycarbonyl-9-methyl-Δ4(10)-octalin for formal total syntheses of certain sesquiterpenoids and diterpenoids. Chem. Pharm. Bull. 1989, 37, 712–717. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.K.; Pita-Boente, M.I. Synthetic approaches to glutinosone. Recl. Trav. Chim. Pays-Bas 1989, 108, 408–412. [Google Scholar] [CrossRef]
- Darvesh, S.; Grant, A.S.; MaGee, D.I.; Valenta, Z. An approach to the synthesis of bruceantin. The synthesis of a tetracyclic intermediate. Can. J. Chem. 1989, 67, 2237–2240. [Google Scholar] [CrossRef]
- Darvesh, S.; Grant, A.S.; MaGee, D.I.; Valenta, Z. Synthetic studies towards bruceantin. Part 1. Establishment of the carbon network. Can. J. Chem. 1991, 69, 712–722. [Google Scholar] [CrossRef]
- Jung, M.E.; Gomez, A.V. Efficient method for the preparation of 2α,3β-dichloro-4,4,10-trimethyldecalin systems as a route for the synthesis of dichlorolissoclimide. Tetrahedron Lett. 1993, 34, 2891–2894. [Google Scholar] [CrossRef]
- Jung, M.E.; Duclos, B.A. Synthetic approach to analogues of betulinic acid. Tetrahedron 2006, 62, 9321–9334. [Google Scholar] [CrossRef]
- Burns, D.J.; Mommer, S.; O’Brien, P.; Taylor, R.J.K.; Whitwood, A.C.; Hachisu, S. Stereocontrolled Synthesis of the AB rings of samaderine C. Org. Lett. 2013, 15, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xing, Z.; Liu, L.; Zhang, H.; Zhong, Z.; Xie, D.X.; She, X. Concise total synthesis of isospongian diterpenoid (±)-polyrhaphin D. ChemistrySelect 2016, 1, 2225–2227. [Google Scholar] [CrossRef]
- Kim, D.E.; Zweig, J.E.; Newhouse, T.R. Total synthesis of paspaline A and emindole PB enabled by computational augmentation of a transform-guided retrosynthetic strategy. J. Am. Chem. Soc. 2019, 141, 1479–1483. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Oritani, T.; Yamashita, K. Enantioselective synthesis of (+)-methyl trisporate B. Agric. Biol. Chem. 1987, 51, 2291–2293. [Google Scholar]
- Takahashi, S.; Oritani, T.; Yamashita, K. Total synthesis of (+)-methyl trisporate B, fungal sex hormone. Tetrahedron 1988, 44, 7081–7088. [Google Scholar] [CrossRef]
- Hagiwara, H.; Uda, H. Optically pure (4aS)-(+)- or (4aR)-(‒)-1,4a-dimethyl-4,4a,7,8-tetrahydronaphtalene-2,5(3H,6H)-dione and its use in the synthesis of an inhibitor of steroid biosynthesis. J. Org. Chem. 1988, 53, 2308–2311. [Google Scholar] [CrossRef]
- Hagiwara, H.; Uda, H. Total synthesis of (+)-dysideapalaunic acid. J. Chem. Soc. Chem. Commun. 1988, 815–817. [Google Scholar] [CrossRef]
- Hagiwara, H.; Uda, H. Total synthesis and absolute stereostructure of (+)-dysideapalaunic acid. J. Chem. Soc. Perkin Trans. 1 1991, 1803–1807. [Google Scholar] [CrossRef]
- Bruner, S.D.; Radeke, H.S.; Tallarico, J.A.; Snapper, M.L. Total synthesis of (‒)-ilimaquinone. J. Org. Chem. 1995, 60, 1114–1115. [Google Scholar] [CrossRef]
- Hagiwara, H.; Inome, K.; Uda, H. A total synthesis of an antibacterial clerodane, 16-hydroxycleroda-3,13(14)-Z-dien-l5,16-olide. J. Chem. Soc. Perkin Trans. 1 1995, 757–764. [Google Scholar] [CrossRef]
- Locke, E.P.; Hecht, S.M. Enantiospecific total synthesis of (+)- and (‒)-avarone and –avarol. Chem. Commun. 1996, 2717–2718. [Google Scholar] [CrossRef]
- Kawano, H.; Itoh, M.; Katoh, T.; Terashima, S. Studies toward the synthesis of popolohuanone E: Synthesis of natural (+)-arenarol related to the proposed biogenetic precursor of popolohuanone E. Tetrahedron Lett. 1997, 38, 7769–7772. [Google Scholar] [CrossRef]
- Corey, E.J.; Roberts, B.E. Total synthesis of dysidiolide. J. Am. Chem. Soc. 1997, 119, 12425–12431. [Google Scholar] [CrossRef]
- Poigny, S.; Guyot, M.; Samadi, M. Efficient total synthesis of (‒)-ilimaquinone. J. Org. Chem. 1998, 63, 5890–5894. [Google Scholar] [CrossRef]
- Xiang, A.X.; Watson, D.A.; Ling, T.; Theodorakis, E.A. Total synthesis of clerocidin via a novel, enantioselective homoallenylboration methodology. J. Org. Chem. 1998, 63, 6774–6775. [Google Scholar] [CrossRef]
- Almstead, J.I.K.; Demuth, T.P., Jr.; Ledoussal, B. An investigation into the total synthesis of clerocidin: Stereoselective synthesis of a clerodane intermediate. Tetrahedron Asymmetry 1998, 9, 3179–3183. [Google Scholar] [CrossRef]
- Hagiwara, H.; Nagatomo, H.; Kazayama, S.; Sakai, H.; Hoshi, T.; Suzuki, T.; Ando, M. Total synthesis of a monocyclofarnesane norsesquiterpenoid isolated from mushroom ingested by beetle: Utility of solid state Bayer-Villiger oxidation. J. Chem. Soc. Perkin Trans. 1 1999, 457–459. [Google Scholar] [CrossRef]
- Ling, T.; Xiang, A.X.; Theodorakis, E.A. Enantioselective total synthesis of avarol and avarone. Angew. Chem. Int. Ed. 1999, 38, 3089–3091. [Google Scholar] [CrossRef]
- Markó, I.E.; Wiaux, M.; Warriner, S.M.; Giles, P.R.; Eustace, P.; Dean, D.; Bailey, M. Towards the total synthesis of clerocidin. Efficient assembly of decalin subunit. Tetrahedron Lett. 1999, 40, 5629–5632. [Google Scholar] [CrossRef]
- Kende, A.S.; Rustenhoven, J.J.; Zimmermann, K. Total synthesis of 15-deoxoclerocidin. Tetrahedron Lett. 2000, 41, 843–846. [Google Scholar] [CrossRef]
- Hagiwara, H.; Nagatomo, H.; Yoshii, F.; Hoshi, T.; Suzuki, T.; Ando, M. Total synthesis of a monocyclofarnesane dinorsesquiterpenoid isolated from mushroom ingested by beetle: Selectivity in solid-state Baeyer-Villiger reaction. J. Chem. Soc. Perkin Trans. 1 2000, 2645–2648. [Google Scholar] [CrossRef]
- Stahl, P.; Kissau, L.; Mazitschek, R.; Huwe, A.; Furet, P.; Giannis, A.; Waldmann, H. Total synthesis and biological evaluation of the nakijiquinones. J. Am. Chem. Soc. 2001, 123, 11586–11593. [Google Scholar] [CrossRef] [PubMed]
- Katoh, T.; Nakatani, M.; Shikita, S.; Sampe, R.; Ishiwata, A.; Ohmori, O.; Nakamura, M.; Terashima, S. Studies toward the total synthesis of popolohuanone E: Enantioselective synthesis of 8-O-methylpopolohuanone E. Org. Lett. 2001, 3, 2701–2704. [Google Scholar] [CrossRef] [PubMed]
- Assefa, H.; Nimrod, A.; Walker, L.; Sindelar, R. Enantioselective synthesis and complement inhibitory assay of A/B- ring partial analogues of oleanolic acid. Bioorg. Med. Chem. Lett. 2001, 11, 1619–1623. [Google Scholar] [CrossRef]
- Cheung, A.K.; Snapper, M.L. Total syntheses of (+)- and (-)-cacospongionolide B: New insight into structural requirements for phospholipase A2 inhibition. J. Am. Chem. Soc. 2002, 124, 11584–11585. [Google Scholar] [CrossRef]
- Favaloro, F.G.; Honda, T.; Honda, Y.; Gribble, G.W.; Suh, N.; Risingsong, R.; Sporn, M.B. Design and synthesis of tricyclic compounds with enone functionalities in rings A and C: A novel class of highly active inhibitors of nitric oxide production in mouse macrophages. J. Med. Chem. 2002, 45, 4801–4805. [Google Scholar] [CrossRef]
- Ling, T.; Rivas, F.; Theodorakis, E.A. Stereoselective synthesis of the fully functionalized core fragment of terpentecin. Tetrahedron Lett. 2002, 43, 9019–9022. [Google Scholar] [CrossRef]
- Ling, T.; Poupon, E.; Rueden, E.J.; Kim, S.H.; Theodorakis, E.A. Unified synthesis of quinone sesquiterpenes based on a radical decarboxylation and quinone addition reaction. J. Am. Chem. Soc. 2002, 124, 12261–12267. [Google Scholar] [CrossRef]
- Iwasaki, K.; Nakatani, M.; Inoue, M.; Katoh, T. Studies toward the total synthesis of (‒)-kampanol A: An efficient construction of the ABCD ring system. Tetrahedron Lett. 2002, 43, 7937–7940. [Google Scholar] [CrossRef]
- Ling, T.; Poupon, E.; Rueden, E.J.; Theodorakis, E.A. Synthesis of (‒)-ilimaquinone via a radical decarboxylation and quinone addition reaction. Org. Lett. 2002, 4, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, M.; Nakamura, M.; Suzuki, A.; Inoue, M.; Katoh, T. A new strategy toward the total synthesis of stachyflin, a potent anti-influenza a virus agent: Concise route to the tetracyclic core structure. Org. Lett. 2002, 4, 4483–4486. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Suzuki, A.; Nakatani, M.; Fuchikami, T.; Inoue, M.; Katoh, T. An efficient synthesis of (+)-aureol via boron trifluoride etherate-promoted rearrangement of (+)-arenarol. Tetrahedron Lett. 2002, 43, 6929–6932. [Google Scholar]
- Zhang, F.; Danishefsky, S.J. An efficient stereoselective total synthesis of dl-sesquicillin, a glucocorticoid antagonist. Angew. Chem. Int. Ed. 2002, 41, 1434–1437. [Google Scholar] [CrossRef]
- Honda, T.; Favaloro, F.G.; Janosik, T.; Honda, Y.; Suh, N.; Sporn, M.B.; Gribble, G.W. Efficient synthesis of (‒)- and (+)-tricyclic compounds with enone functionalities in rings A and C. A novel class of orally active anti-inflammatory and cancer chemopreventive agents. Org. Biomol. Chem. 2003, 1, 4384–4391. [Google Scholar] [CrossRef]
- Iwasaki, K.; Nakatani, M.; Inoue, M.; Katoh, T. Synthetic studies of kampanols, novel p21ras farnesyltransferase inhibitors: An efficient synthesis of the tetracyclic ABCD ring system of kampanols. Tetrahedron 2003, 59, 8763–8773. [Google Scholar] [CrossRef]
- Suzuki, A.; Nakatani, M.; Nakamura, M.; Kawaguchi, K.; Inoue, M.; Katoh, T. Highly improved synthesis of (+)-aureol via (-)-neoavarone and (-)-neoavarol, by employing salcomine oxidation and acid-induced rearrangement/cyclization strategy. Synlett 2003, 329–332. [Google Scholar] [CrossRef]
- Cheung, A.K.; Murelli, R.; Snapper, M.L. Total syntheses of (+)- and (‒)-cacospongionolide B, cacospongionolide E, and related analogues. Preliminary study of structural features required for phospholipase A2 inhibition. J. Org. Chem. 2004, 69, 5712–5719. [Google Scholar] [CrossRef]
- Guo, Z.; Vangapandu, S.; Nimrod, A.; Walker, L.A.; Sindelar, R.D. Synthesis of A/B-ring partial analogs of bruceantin as potential antimalarial agents. Med. Chem. 2005, 1, 3–11. [Google Scholar] [CrossRef]
- Hagiwara, H.; Hamano, K.; Nozawa, M.; Hoshi, T.; Suzuki, T.; Kido, F. The first total synthesis of (-)-methyl barbascoate. J. Org. Chem. 2005, 70, 2250–2255. [Google Scholar] [CrossRef]
- Watanabe, K.; Iwasaki, K.; Abe, T.; Inoue, M.; Ohkubo, K.; Suzuki, T.; Katoh, T. Enantioselective total synthesis of (-)-candelalide A, a novel blocker of the voltage-gated potassium channel Kv1.3 for an immunosuppressive agent. Org. Lett. 2005, 7, 3745–3748. [Google Scholar] [CrossRef] [PubMed]
- Takikawa, H.; Imamura, Y.; Sasaki, M. Synthesis and absolute configuration of brevione B, an allelochemical isolated from Penicillium sp. Tetrahedron 2006, 62, 39–48. [Google Scholar] [CrossRef]
- Murelli, R.P.; Cheung, A.K.; Snapper, M.L. Conformationally restricted (+)-cacospongionolide B analogues. Influence on secretory phospholipase A2 inhibition. J. Org. Chem. 2007, 72, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, M.; Suka, Y.; Hoshi, T.; Suzuki, T.; Hagiwara, H. Total synthesis of the hallucinogenic neoclerodane diterpenoid salvinorin A. Org. Lett. 2008, 10, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, J.; Oguchi, T.; Watanabe, K.; Abe, H.; Kanno, S.; Ishikawa, M.; Katoh, T. Highly efficient total synthesis of the marine natural products (+)-avarone, (+)-avarol, (−)-neoavarone, (−)-neoavarol and (+)-aureol. Chem. Eur. J. 2008, 14, 829–837. [Google Scholar] [CrossRef]
- Scheck, M.; Koch, M.A.; Waldmann, H. Synthesis of a dysidiolide-inspired compound library and discovery of acetylcholinesterase inhibitors based on protein structure similarity clustering (PSSC). Tetrahedron 2008, 64, 4792–4802. [Google Scholar] [CrossRef]
- Oguchi, T.; Watanabe, K.; Ohkubo, K.; Abe, H.; Katoh, T. Enantioselective total synthesis of (-)-candelalides A, B and C: Potential Kv1.3 blocking immunosuppressive agents. Chem. Eur. J. 2009, 15, 2826–2845. [Google Scholar] [CrossRef]
- Oshida, M.; Ono, M.; Nakazaki, A.; Kobayashi, S. Total synthesis of (+)-cacospongionolide B. Heterocycles 2010, 80, 313–328. [Google Scholar]
- Pereira, A.R.; Strangman, W.K.; Marion, F.; Feldberg, L.; Roll, D.; Mallon, R.; Hollander, I.; Andersen, R.J. Synthesis of phosphatidylinositol 3-kinase (PI3K) inhibitory analogues of the sponge meroterpenoid liphagal. J. Med. Chem. 2010, 53, 8523–8533. [Google Scholar] [CrossRef]
- Macías, F.A.; Carrera, C.; Chinchilla, N.; Fronczek, F.R.; Galindo, J.C.G. Synthesis of the western half of breviones C, D, F and G. Tetrahedron 2010, 66, 4125–4132. [Google Scholar] [CrossRef]
- Watanabe, K.; Sakurai, J.; Abe, H.; Katoh, T. Total synthesis of (+)-stachyflin: A potential anti-influenza A virus agent. Chem. Commun. 2010, 46, 4055–4057. [Google Scholar] [CrossRef] [PubMed]
- Ling, T.; Xu, J.; Smith, R.; Ali, A.; Cantrell, C.L.; Theodorakis, E.A. Synthesis of (‒)-callicarpenal, a potent arthropod repellent. Tetrahedron 2011, 67, 3023–3029. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, J.; Kikuchi, T.; Takahashi, O.; Watanabe, K.; Katoh, T. Enantioselective total synthesis of (+)-stachyflin: A potential anti-influenza. A virus agent isolated from a microorganism. Eur. J. Org. Chem. 2011, 2948–2957. [Google Scholar] [CrossRef]
- Schmalzbauer, B.; Herrmann, J.; Muller, R.; Menche, D. Total synthesis and antibacterial activity of dysidavarone A. Org. Lett. 2013, 15, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, H.; Honma, N.; Kinugawa, K.; Sato, S.; Hoshi, T.; Suzuki, T. Second generation synthesis of the neo-clerodane diterpenoid methyl barbascoate. Nat. Prod. Commun. 2013, 8, 873–875. [Google Scholar] [CrossRef] [Green Version]
- Fukui, Y.; Narita, K.; Katoh, T. Enantioselective total synthesis of dysidavarone A, a novel sesquiterpenoid quinone from the marine sponge Dysidea avara. Chem. Eur. J. 2014, 20, 2436–2439. [Google Scholar] [CrossRef]
- Wan, K.K.; Iwasaki, K.; Umotoy, J.C.; Wolan, D.W.; Shenvi, R.A. Nitrosopurines en route to potently cytotoxic asmarines. Angew. Chem. Int. Ed. 2015, 54, 2410–2415. [Google Scholar] [CrossRef] [Green Version]
- Sumii, Y.; Kotoku, N.; Fukuda, A.; Kawachi, T.; Sumii, Y.; Arai, M.; Kobayashi, M. Enantioselective synthesis of dictyoceratin-A (smenospondiol) and –C, hypoxia-selective growth inhibitors from marine sponge. Bioorg. Med. Chem. 2015, 23, 966–975. [Google Scholar] [CrossRef]
- Lu, J.; Aguilar, A.; Zou, B.; Bao, W.; Koldas, S.; Shi, A.; Desper, J.; Wangemann, P.; Xie, X.S.; Hua, D.H. Chemical synthesis of tetracyclic terpenes and evaluation of antagonistic activity on endothelin-A receptors and voltage-gated calcium channels. Bioorg. Med. Chem. 2015, 23, 5985–5998. [Google Scholar] [CrossRef] [Green Version]
- Dhiman, S.; Ramasastry, S.S.V. One-pot relay gold(I) and Brønsted acid catalysis: Cyclopenta[b]annulation of indoles via hydroamination/Nazarov-type cyclization cascade of enynols. Org. Lett. 2015, 17, 5116–5119. [Google Scholar] [CrossRef]
- Dethe, D.H.; Sau, S.K.; Mahapatra, S. Biomimetic enantioselective total synthesis of (‒)-mycoleptodiscin. Org. Lett. 2016, 18, 6392–6395. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zhang, X.; Zhang, J.; Shen, Z. Total synthesis of dysidavarone A. Tetrahedron 2016, 72, 4337–4345. [Google Scholar] [CrossRef]
- Werner, B.; Kalesse, M. Pinacol coupling strategy for the construction of the bicyclo[6.4.1]tridecane framework of schiglautone A. Org. Lett. 2017, 19, 1524–1526. [Google Scholar] [CrossRef] [PubMed]
- Swapnil, N.; Kumar, A.S.; Babu, N.J.; Yadav, J.S. Synthetic approach towards the synthesis of antimalarial gomphostenin. Asian J. Org. Chem. 2017, 6, 1091–1098. [Google Scholar] [CrossRef]
- Pinkerton, D.M.; Vanden Berg, T.J.; Bernhardt, P.V.; Williams, C.M. Gaining synthetic appreciation for the gedunin ABC ring system. Chem. Eur. J. 2017, 23, 2282–2285. [Google Scholar] [CrossRef] [Green Version]
- Pinkerton, D.M.; Bernhardt, P.V.; Savage, G.P.; Williams, C.M. Towards the total synthesis of gedunin: Construction of the fully elaborated ABC ring system. Asian J. Org. Chem. 2017, 6, 583–597. [Google Scholar] [CrossRef]
- He, C.; Hu, J.; Wu, Y.; Ding, H. Total syntheses of highly oxidized ent-kaurenoids pharicin A, pharicinin B, 7-O-acetylpseurata C, and pseurata C: A [5+2] cascade approach. J. Am. Chem. Soc. 2017, 139, 6098–6101. [Google Scholar] [CrossRef]
- Dethe, D.H.; Sau, S.K. Biomimetic enantioselective total synthesis of (‒)-petromindole. Org. Lett. 2018, 20, 632–635. [Google Scholar] [CrossRef]
- Dethe, D.H.; Mahapatra, S.; Sau, S.K. Enantioselective total synthesis and assignment of the absolute configuration of the meroterpenoid (+)-taondiol. Org. Lett. 2018, 20, 2766–2769. [Google Scholar] [CrossRef]
- Haque, M.A.; Sailo, B.L.; Padmavathi, G.; Kunnumakkara, A.B.; Jana, C.K. Nature-inspired development of unnatural meroterpenoids as the non-toxic anti-colon cancer agents. Eur. J. Med. Chem. 2018, 160, 256–265. [Google Scholar] [CrossRef]
- Dethe, D.H.; Sau, S.K. Total synthesis of (+)-strongylophorines 2 and 9. Org. Lett. 2019, 21, 3799–3803. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Srinivasan, K.G.; Venkataramani, P.S. Rearrangements of bicyclic-δ-hydroxy-α,β-enones-IV. Tetrahedron 1970, 26, 1453–1461. [Google Scholar] [CrossRef]
- Dutcher, J.S.; Macmillan, J.G.; Heathcock, C.H. Pentacyclic triterpene synthesis. II. Preparation of an AB synthon. Tetrahedron Lett. 1974, 15, 929–932. [Google Scholar] [CrossRef]
- Snitman, D.L.; Tsai, M.-Y.; Watt, D.S.; Edwards, C.L.; Stotter, P.L. Convenient syntheses of 5,5,9-trimethyl-trans-1-decalone and 6β-hydroxy-5,5,9β-trimethyl-trans-1-decalone. J. Org. Chem. 1979, 44, 2838–2842. [Google Scholar] [CrossRef]
- Fuhshuku, K.; Tomita, M.; Sugai, T. Enantiomerically pure octahydronaphtalenone and octahydroindenone: Elaboration of the substrate overcame the specificity of yeast-mediated reduction. Adv. Synth. Catal. 2003, 345, 766–774. [Google Scholar] [CrossRef]
- Takatori, K.; Nakayama, M.; Futaishi, N.; Yamada, S.; Hirayama, S.; Kajiwara, M. Solid-supported Robinson annulation under microwave irradiation. Chem. Pharm. Bull. 2003, 51, 455–457. [Google Scholar] [CrossRef] [Green Version]
- Kawanami, H.; Ikushima, Y. Promotion of one-poi Robinson annelation achieved by gradual pressure and temperature manipulation under supercritical conditions. Tetrahedron Lett. 2004, 45, 5147–5150. [Google Scholar] [CrossRef]
- Eder, U.; Sauer, G.; Wiechert, R. New type of asymmetric cyclization to optically active steroid CD partial structures. Angew. Chem. Int. Ed. 1971, 10, 496–497. [Google Scholar] [CrossRef]
- Hiroi, K.; Yamada, S.I. Stereochemical studies. XXXVIII. Asymmetric synthesis of the key compounds for the synthesis of optically active diterpenes. Asymmetric synthesis of optically active 1,2,3,4,5,6,7,8,8a-octahydro-8a-methyl-3,8-naphtalenedione derivatives with l-proline derivatives. Chem. Pharm. Bull. 1975, 23, 1103–1109. [Google Scholar]
- Uma, R.; Swaminathan, S.; Rajagopalan, K. Base catalyzed rearrangement of oxy-Cope systems. Tetrahedron Lett. 1984, 25, 5825–5828. [Google Scholar] [CrossRef]
- Uma, R.; Rajagopalan, K.; Swaminathan, S. Base catalysed rearrangement of oxy-Cope systems. Tetrahedron Lett. 1986, 42, 2757–2769. [Google Scholar] [CrossRef]
- Leonelli, F.; Garofalo, B.; Migneco, L.M.; Marini Bettolo, R.; Colais, F.; Sinibaldi, M. Chiral HPLC Resolution of the Wieland-Miescher Ketone and Derivatives. J. Liq. Cromatogr. Relat. Technol. 2003, 3, 409–424. [Google Scholar] [CrossRef]
- Shigehisa, H.; Mizutani, T.; Tosaki, S.; Ohshima, T.; Shibasaki, M. Formal total synthesis of (+)-wortmannin using catalytic asymmetric intramolecular aldol condensation reaction. Tetrahedron 2005, 61, 5057–5065. [Google Scholar] [CrossRef]
- Nozawa, M.; Akita, T.; Hoshi, T.; Suzuki, T.; Hagiwara, H. Recyclable asymmetric cyclization in ionic liquid catalysed by an amino acid, leading to a Wieland-Miescher ketone analogue. Synlett 2007, 4, 661–663. [Google Scholar]
- Nagamine, T.; Inomata, K.; Endo, Y.; Paquette, L.A. Amino acid mediated intramolecular asymmetric aldol reaction to construct a new chiral bicyclic enedione containing a seven-membered ring: Remarkable inversion of enantioselectivity compared to the six-membered ring example. J. Org. Chem. 2007, 72, 123–131. [Google Scholar] [CrossRef]
- Lanfranchi, D.A.; Baldovini, N.; Hanquet, G. Large-scale preparation of enantiomerically pure (4aR)-(‒)-1,4a-dimethyl-4,4a,7,8-tetrahydronaphthalene-2,5(3H,6H)-dione: A useful Wieland-Miescher diketone analogue. Synthesis 2008, 23, 3775–3778. [Google Scholar]
- Carrera, C.; Chichilla, N.; Fronczek, F.R.; Galindo, J.C.G.; Macías, F.A. Structure-activity relationship studies of the phytotoxic properties of the diterpenic moiety of breviones. Pest. Manag. Sci. 2015, 71, 701–711. [Google Scholar] [CrossRef]
- Cañellas, S.; Ayats, C.; Henseler, A.H.; Pericàs, M.A. A highly active polymer-supported catalyst for asymmetric Robinson annulations in continuous flow. ACS Catal. 2017, 7, 1383–1391. [Google Scholar] [CrossRef]
- Schiavo, L.; Lebedel, L.; Massè, P.; Choppin, S.; Hanquet, G. Access to Wieland-Miescher diketone-derived building blocks by stereoselective construction of the C-9 quaternary carbon center using the Mukaiyama aldol reaction. J. Org. Chem. 2018, 83, 6247–6258. [Google Scholar] [CrossRef]
- Kawai, N.; Takao, K.; Kobayashi, S. Synthetic study of Akaterpin: Determination of the relative stereochemistry of the upper decalin moiety with disulfated hydroquinone. Tetrahedron Lett. 1999, 40, 4193–4196. [Google Scholar] [CrossRef]
- Schiavo, L.; Jeanmart, L.; Lanners, S.; Choppin, S.; Hanquet, G. FeCl3 6H2O/acetaldehyde, a versatile system for the deprotection of ketals and acetals via a transacetalization process. New J. Chem. 2017, 41, 1421–1424. [Google Scholar] [CrossRef]
- Ardon-Jimenez, A.; Halsall, T.G. The reactions of 5a-Allyl-1,1-ethylenedioxy-5β,9β-dimethyl-trans-decalin-6-one, a potential intermediate in the synthesis of friedolabdanes. J. Chem. Soc. Perkin Trans. 1 1978, 1461–1470. [Google Scholar] [CrossRef]
- Snitman, D.L.; Tsai, M.Y.; Watt, D.S. Robinson annulations of sterically hindered α-carbomethoxycyclohexanones. Synth. Commun. 1978, 8, 195–204. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Pita Boente, M.I. A synthetic approach to noreudesmanoid phytoalexins. Heterocycles 1985, 23, 5–10. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Carrasco, M.C.; Peña-Matheud, C.A. Observations on the dehydrogenation of α,β-unsaturated carbonyl compounds with thallium(III) acetate. Recl. Trav. Chim. Pays-Bas 1989, 108, 94–96. [Google Scholar] [CrossRef]
- Haque, M.A.; Jana, C.K. Regiodivergent remote arylation of cycloalkanols to dysideanone’s fused carbotetracycles and its bridged isomers. Chem. Eur. J. 2017, 23, 13300–13304. [Google Scholar] [CrossRef]
- Brufani, M.; Ceccacci, F.; Filocamo, L.; Garofalo, B.; Joudioux, R.; La Bella, A.; Leonelli, F.; Migneco, L.M.; Marini Bettolo, R.; Farina, P.M.; et al. Novel locally active estrogens accelerate cutaneous wound healing. A preliminary study. Mol. Pharm. 2009, 6, 543–556. [Google Scholar] [CrossRef]
- Leonelli, F.; Blesi, F.; Dirito, P.; Trombetta, A.; Ceccacci, F.; La Bella, A.; Migneco, L.M.; Marini Bettolo, R. Diastereoselective total synthesis of (+)-13-stemarene by fourth generation methods: A formal total synthesis of (+)-18-deoxystemarin. J. Org. Chem. 2011, 76, 6871–6876. [Google Scholar] [CrossRef]
- Leonelli, F.; Latini, V.; Trombetta, A.; Bartoli, G.; Ceccacci, F.; La Bella, A.; Sferrazza, A.; Lamba, D.; Migneco, L.M.; Marini Bettolo, R. Regio- and diastereoselective synthesis and X-ray structure determination of (+)-2-deoxyoryzalexin S from (+)-podocarpic acid. Structural nonidentity with its nominal natural isolated enantiomer. J. Nat. Prod. 2012, 75, 1944–1950. [Google Scholar] [CrossRef]
- Petaccia, M.; Condello, M.; Giansanti, L.; La Bella, A.; Leonelli, F.; Meschini, S.; Gradella Villalva, D.; Pellegrini, E.; Ceccacci, F.; Galantini, L.; et al. Inclusion of new 5-fluorouracil amphiphilic derivatives in liposome formulation for cancer treatment. Med. Chem. Commun. 2015, 6, 1639–1642. [Google Scholar] [CrossRef]
- La Bella, A.; Leonelli, F.; Migneco, L.M.; Marini Bettolo, R. (+)-Podocarpic acid as chiral template in the synthesis of aphidicolane, stemodane and stemarane diterpenoids. Molecules 2016, 21, 1197. [Google Scholar] [CrossRef] [Green Version]
- Leonelli, F.; Mostarda, A.; De Angelis, L.; Lamba, D.; Demitri, N.; La Bella, A.; Ceccacci, F.; Migneco, L.M.; Marini Bettolo, R. Proof of the structure of the Stemodia chilensis tetracyclic diterpenoid (+)-19-acetoxystemodan-12-ol by synthesis from (+)-podocarpic acid: X-ray structure determination of a key intermediate. J. Nat. Prod. 2016, 79, 1155–1159. [Google Scholar] [CrossRef]
- Leonelli, F.; Trombetta, A.; La Bella, A.; Lucarelli, G.; Demitri, N.; Lamba, D.; Migneco, L.M.; Marini Bettolo, R. Enantioselective synthesis and X-ray structure of (+)((4aS,5S,8aS)-5,8a-dimethyl-7-methyleneoctahydro-2Hspiro[naphthalene-1,2′-[1,3]dioxolan]-5-yl)methyl-4-iodobenzoate. Eur. J. Org. Chem. 2019, 1594–1599. [Google Scholar] [CrossRef]
- Leonelli, F.; Valletta, A.; Migneco, L.M.; Marini Bettolo, R. Stemarane diterpenes and diterpenoids. Int. J. Mol. Sci. 2019, 20, 2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciceri, P.; Demnitz, F.W.J. An efficient, rapid and highly selective preparation of the Wieland-Miescher ketone-9-ethylene ketal. Tetrahedron Lett. 1997, 38, 389–390. [Google Scholar] [CrossRef]
- Van Gool, M.; Vandewalle, M. Vitamin D: Enantioselective synthesis of (3aR,4R,7aS)-4-hydroxy-7amethylperhydro-1-indenone, a suitable CD-ring fragment. Eur. J. Org. Chem. 2000, 3427–3431. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Reaction Time (h) | TsOH (mmol) | 1,2-Ethanediol (mmol) | (+)-5 ee (%) 2 | (+)-1 ee (%) 2 | (+)-1 (%) 3 | H2O (mmol) 4 |
|---|---|---|---|---|---|---|---|
| 1 | 2.5 | 0.2 | 1.2 | 60 | 80 | 7 | - |
| 2 | 5 | 0.2 | 1.2 | 30 | 66 | 2 | - |
| 3 | 24 | 0.2 | 1.2 | 10 | 60 | 1 | - |
| 4 | 2.5 | 0.01 | 14.5 | 96 | 96 | 4 | - |
| 5 | 2.5 | 0.2 | - | - | 96 | 100 | - |
| 6 | 2.5 | 0.2 | - | - | 96 | 100 | 1 |
| 7 | 2.5 | 0.2 | - | - | 96 | 100 | 53 |
| 8 | 2.5 | 0.2 | - | - | 96 | 100 | 53 5 |
| Entry | TsOH (mmol) | 1,2-Ethanediol (mmol) | (+)-5 ee (%) 2 | (+)-1 ee (%) 2 | (+)-1 (%) 3 |
|---|---|---|---|---|---|
| 1 | - | 1.2 | 96 | - | 0 |
| 2 | 0.2 | 1.2 | 12 | - | 0 |
| 3 | 0.2 | - | 34 | 70 | 10 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leonelli, F.; Piergentili, I.; Lucarelli, G.; Migneco, L.M.; Marini Bettolo, R. Unexpected Racemization in the Course of the Acetalization of (+)-(S)-5-Methyl-Wieland–Miescher Ketone with 1,2-Ethanediol and TsOH under Classical Experimental Conditions. Int. J. Mol. Sci. 2019, 20, 6147. https://doi.org/10.3390/ijms20246147
Leonelli F, Piergentili I, Lucarelli G, Migneco LM, Marini Bettolo R. Unexpected Racemization in the Course of the Acetalization of (+)-(S)-5-Methyl-Wieland–Miescher Ketone with 1,2-Ethanediol and TsOH under Classical Experimental Conditions. International Journal of Molecular Sciences. 2019; 20(24):6147. https://doi.org/10.3390/ijms20246147
Chicago/Turabian StyleLeonelli, Francesca, Irene Piergentili, Giulio Lucarelli, Luisa Maria Migneco, and Rinaldo Marini Bettolo. 2019. "Unexpected Racemization in the Course of the Acetalization of (+)-(S)-5-Methyl-Wieland–Miescher Ketone with 1,2-Ethanediol and TsOH under Classical Experimental Conditions" International Journal of Molecular Sciences 20, no. 24: 6147. https://doi.org/10.3390/ijms20246147
APA StyleLeonelli, F., Piergentili, I., Lucarelli, G., Migneco, L. M., & Marini Bettolo, R. (2019). Unexpected Racemization in the Course of the Acetalization of (+)-(S)-5-Methyl-Wieland–Miescher Ketone with 1,2-Ethanediol and TsOH under Classical Experimental Conditions. International Journal of Molecular Sciences, 20(24), 6147. https://doi.org/10.3390/ijms20246147