Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpen Aminodiols



Abstract
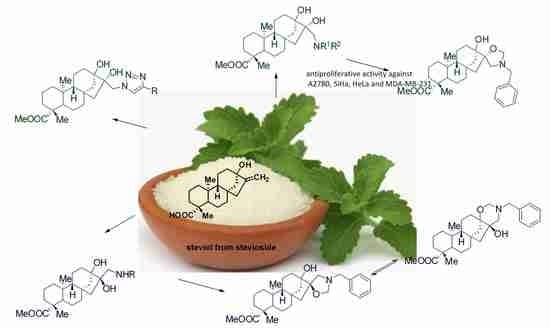
:
1. Introduction
2. Results
2.1. Synthesis of Steviol-Based Epoxyalcohol Key Intermediates 4 and 5
2.2. Synthesis of Steviol-Based Dihydroxytriazoles via Azidodiol 6
2.3. Synthesis of Steviol-Based Aminodiol Derivatives
2.4. Application of Aminodiol Derivatives as Chiral Ligands for Catalytic Addition of Diethylzinc to Benzaldehyde
2.5. Antiproliferative Activity of Aminodiols
3. Discussion
4. Materials and Methods
4.1. General Methods
4.2. Starting Materials
4.3. Epoxydation of Steviol Methyl Ester 3
4.3.1. (2’S,4R,6aS,9S,11aR,11bS)-Methyl 9-hydroxy-4,11b-dimethyldodecahydro-1H-spiro[6a,9-methanocyclohepta[a]naphthalene-8,2’-oxirane]-4-carboxylate (4)
4.3.2. (2’R,4R,6aS,9S,11aR,11bS)-Methyl9-hydroxy-4,11b-dimethyldodecahydro-1H-spiro[6a,9-methanocyclohepta[a]naphthalene-8,2’-oxirane]-4-carboxylate (5)
4.4. (4R,6aS,8S,9S,11aR,11bS)-Methyl8-(azidomethyl)-8,9-dihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (6)
4.5. General Procedure for the “Click” Reaction of 6 for the Preparation of 7–10
4.5.1. (4R,6aS,8S,9S,11aR,11bS)-Methyl 8,9-dihydroxy-4,11b-dimethyl-8-((4-phenyl-1H-1,2,3-triazol-1-yl) methyl) tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (7)
4.5.2. Cyclopenta-2,4-dien-1-yl(2-(1-(((4R,6aS,8S,9S,11aR,11bS)-8,9-dihydroxy-4-(methoxycarbonyl)-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalen-8-yl)methyl)-1H-1,2,3-triazol-4-yl)cyclopenta-2,4-dien-1-yl)iron (8)
4.5.3. (4R,6aS,8S,9S,11aR,11bS)-Methyl 8,9-dihydroxy-4,11b-dimethyl-8-((4-(pyridin-2-yl)-1H-1,2,3-triazol-1-yl) methyl) tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (9)
4.5.4. (4R,6aS,8S,9S,11aR,11bS)-Methyl8-((4-cyclopropyl-1H-1,2,3-triazol-1-yl)methyl)-8,9-dihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (10)
4.6. General Procedure for the Preparation of 11–18
4.6.1. (4R,6aS,8S,9S,11aR,11bS)-Methyl 8-((benzylamino)methyl)-8,9-dihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (11)
4.6.2. (4R,6aS,8S,9S,11aR,11bS)-Methyl 8-((benzyl(methyl)amino)methyl)-8,9-dihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (12)
4.6.3. (4R,6aS,8S,9S,11aR,11bS)-Methyl 8,9-dihydroxy-4,11b-dimethyl-8-((((R)-1-phenylethyl)amino) methyl) tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (13)
4.6.4. (4R,6aS,8S,9S,11aR,11bS)-Methyl 8,9-dihydroxy-4,11b-dimethyl-8-((((S)-1-phenylethyl)amino) methyl) tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (14)
4.6.5. (4R,6aS,8S,9S,11aR,11bS)-Methyl 8,9-dihydroxy-8-((isopropylamino)methyl)-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (15)
4.6.6. (4R,6aS,8S,9S,11aR,11bS)-Methyl 8-((diethylamino)methyl)-8,9-dihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate hydrochloride (16)
4.6.7. (4R,6aS,8S,9S,11aR,11bS)-Methyl 8,9-dihydroxy-4,11b-dimethyl-8-((prop-2-yn-1-ylamino)methyl) tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (17)
4.6.8. (4R,6aS,8S,9S,11aR,11bS)-Methyl 8-(((3,5-bis(trifluoromethyl)benzyl)amino)methyl)-8,9-dihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (18)
4.6.9. (4R,6aS,8R,9S,11aR,11bS)-Methyl 8-((benzylamino)methyl)-8,9-dihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (22)
4.7. General Procedure for Ring Closure of 11 and 22 with Formaldehyde
4.7.1. (4R,5’S,6aS,9S,11aR,11bS)-Methyl3’-benzyl-9-hydroxy-4,11b-dimethyldodecahydro-1H-spiro[6a,9-methanocyclohepta[a]naphthalene-8,5’-oxazolidine]-4-carboxylate (19)
4.7.2. (4R,5’R,6aS,9S,11aR,11bS)-Methyl3’-benzyl-9-hydroxy-4,11b-dimethyldodecahydro-1H-spiro[6a,9-methanocyclohepta[a]naphthalene-8,5’-oxazolidine]-4-carboxylate (24A) and (4R,6aR,7aR,11aS,13aR,13bS)-Methyl 9-benzyl-7a-hydroxy-4,13b-dimethylhexadecahydro-6a,11a-methanonaphtho[1’,2’:5,6]cyclohepta[1,2-e][1,3]oxazine-4-carboxylate (24B)
4.8. (4R,6aS,8S,9S,11aR,11bS)-Methyl 8,9-dihydroxy-4,11b-dimethyl-8-((((1-phenethyl-1H-1,2,3-triazol-4-yl)methyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (20)
4.9. General Procedure for Debenzylation of 11 and 22
4.9.1. (4R,6aS,8S,9S,11aR,11bS)-Methyl 8-(aminomethyl)-8,9-dihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (21)
4.9.2. (4R,6aS,8R,9S,11aR,11bS)-Methyl 8-(aminomethyl)-8,9-dihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (23)
4.10. General Procedure for the Reaction of Benzaldehyde with Diethylzinc in the Presence of Chiral Catalysts
4.11. Determination of Antiproliferative Properties
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| Et2O | Diethyl ether |
| EtOH | Ethanol |
| HCHO | Formaldehyde |
| EtOAc | Ethyl acetate |
| t-BuOOH | tert-Butyl hydroperoxide |
| VO(acac)2 | Vanadyl acetylacetonate |
| DCM | Dichloromethane |
| MeCN | Acetonitrile |
| THF | Tetrahydrofuran |
| DMDO | Dimethyldioxirane |
| Et2Zn | Diethylzinc |
References
- El Alami, M.S.I.; El Amrani, M.A.; Agbossou-Niedercorn, F.; Suisse, I.; Mortreux, A. Chiral Ligands Derived from Monoterpenes: Application in the Synthesis of Optically Pure Secondary Alcohols via Asymmetric Catalysis. Chem. Eur. J. 2015, 21, 1398–1413. [Google Scholar] [CrossRef] [PubMed]
- Grajewska, A.; Rozwadowska, M.D. Stereoselective synthesis of cytoxazone and its analogues. Tetrahedron Asymmetry 2007, 18, 803–813. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Tosh, D.K.; Toti, K.S.; Ciancetta, A. Polypharmacology of conformationally locked methanocarba nucleosides. Drug Discov. Today 2017, 22, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Sadler, J.M.; Mosley, S.L.; Dorgan, K.M.; Zhou, Z.S.; Seley-Radtke, K.L. Synthetic strategies toward carbocyclic purine–pyrimidine hybrid nucleosides. Bioorg. Med. Chem. 2009, 17, 5520–5525. [Google Scholar] [CrossRef] [Green Version]
- Wróblewski, A.E.; Głowacka, I.E.; Piotrowska, D.G. 1′-Homonucleosides and their structural analogues: A review. Eur. J. Med. Chem. 2016, 118, 121–142. [Google Scholar] [CrossRef]
- Kleinert, H.; Rosenberg, S.; Baker, W.; Stein, H.; Klinghofer, V.; Barlow, J.; Spina, K.; Polakowski, J.; Kovar, P.; Cohen, J.; et al. Discovery of a peptide-based renin inhibitor with oral bioavailability and efficacy. Science 1992, 257, 1940–1943. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Mohapatra, S.; Yadav, J.S. Practical synthesis of Abbott amino-diol: A core unit of the potent renin inhibitor Zankiren. Tetrahedron 1999, 55, 4763–4768. [Google Scholar] [CrossRef]
- Toribatake, K.; Miyata, S.; Naganawa, Y.; Nishiyama, H. Asymmetric synthesis of optically active 3-amino-1,2-diols from N-acyl-protected allylamines via catalytic diboration with Rh[bis(oxazolinyl)phenyl] catalysts. Tetrahedron 2015, 71, 3203–3208. [Google Scholar] [CrossRef]
- Paraskar, A.S.; Sudalai, A. Enantioselective synthesis of (−)-cytoxazone and (+)-epi-cytoxazone, novel cytokine modulators via Sharpless asymmetric epoxidation and l-proline catalyzed Mannich reaction. Tetrahedron 2006, 62, 5756–5762. [Google Scholar] [CrossRef]
- Tanaka, T.; Yasuda, Y.; Hayashi, M. New Chiral Schiff Base as a Tridentate Ligand for Catalytic Enantioselective Addition of Diethylzinc to Aldehydes. J. Org. Chem. 2006, 71, 7091–7093. [Google Scholar] [CrossRef]
- Koneva, E.A.; Korchagina, D.V.; Gatilov, Y.V.; Genaev, A.M.; Krysin, A.P.; Volcho, K.P.; Tolstikov, A.G.; Salakhutdinov, N.F. New chiral ligands based on (+)-α-pinene. Russ. J. Org. Chem. 2010, 46, 1109–1115. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Gonda, T.; Ötvös, S.B.; Fülöp, F. Stereoselective syntheses and transformations of chiral 1,3-aminoalcohols and 1,3-diols derived from nopinone. Tetrahedron Asymmetry 2014, 25, 1138–1145. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Csőr, Á.; Csámpai, A.; Fülöp, F. Stereoselective Synthesis and Modelling-Driven Optimisation of Carane-Based Aminodiols and 1,3-Oxazines as Catalysts for the Enantioselective Addition of Diethylzinc to Benzaldehyde. Chem. Eur. J. 2016, 22, 7163–7173. [Google Scholar] [CrossRef] [PubMed]
- Szakonyi, Z.; Csillag, K.; Fülöp, F. Stereoselective synthesis of carane-based aminodiols as chiral ligands for the catalytic addition of diethylzinc to aldehydes. Tetrahedron Asymmetry 2011, 22, 1021–1027. [Google Scholar] [CrossRef]
- Tashenov, Y.; Daniels, M.; Robeyns, K.; Van Meervelt, L.; Dehaen, W.; Suleimen, Y.; Szakonyi, Z. Stereoselective Syntheses and Application of Chiral Bi- and Tridentate Ligands Derived from (+)-Sabinol. Molecules 2018, 23, 771. [Google Scholar] [CrossRef] [Green Version]
- Gonda, T.; Szakonyi, Z.; Csámpai, A.; Haukka, M.; Fülöp, F. Stereoselective synthesis and application of tridentate aminodiols derived from (+)-pulegone. Tetrahedron Asymmetry 2016, 27, 480–486. [Google Scholar] [CrossRef]
- Panev, S.; Linden, A.; Dimitrov, V. Chiral aminoalcohols with a menthane skeleton as catalysts for the enantioselective addition of diethylzinc to benzaldehyde. Tetrahedron Asymmetry 2001, 12, 1313–1321. [Google Scholar] [CrossRef]
- Alberola, A.; Andrés, C.; Pedrosa, R. Diastereoselective Ring Opening of 2-Substituted N -Benzyl-4,4, 7α-trimethyl- trans -octahydro-1,3-benzoxazines by Grignard Reagents. Highly Enantioselective Synthesis of Primary Amines. Synlett 1990, 1990, 763–765. [Google Scholar] [CrossRef]
- Andrés, C.; Nieto, J.; Pedrosa, R.; Villamañán, N. Synthesis of Enantiopure Primary Amines by Stereoselective Ring Opening of Chiral Octahydro-1,3-benzoxazines by Grignard and Organoaluminum Reagents. J. Org. Chem. 1996, 61, 4130–4135. [Google Scholar] [CrossRef]
- Pedrosa, R.; Andrés, C.; Nieto, J.; del Pozo, S. Synthesis of Enantiopure 3-Azabicyclo[3.2.0]heptanes by Diastereoselective Intramolecular [2+2] Photocycloaddition Reactions on Chiral Perhydro-1,3-benzoxazines. J. Org. Chem. 2003, 68, 4923–4931. [Google Scholar] [CrossRef]
- Pedrosa, R.; Andrés, C.; Duque-Soladana, J.P.; Rosón, C.D. Regio- and stereoselective 6-exo-trig radical cyclisations onto chiral perhydro-1,3-benzoxazines: Synthesis of enantiopure 3-alkylpiperidines. Tetrahedron Asymmetry 2000, 11, 2809–2821. [Google Scholar] [CrossRef]
- Ding, J.-Y.; Liu, X.-X.; Xiong, D.-M.; Ye, L.-M.; Chao, R.-B. Simultaneous Determination of Thirteen Aminoalcohol-Diterpenoid Alkaloids in the Lateral Roots of Aconitum carmichaeli by Solid-Phase Extraction-Liquid Chromatography–Tandem Mass Spectrometry. Planta Med. 2014, 80, 723–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dank, C.; Sanichar, R.; Choo, K.-L.; Olsen, M.; Lautens, M. Recent Advances Towards Syntheses of Diterpenoid Alkaloids. Synthesis 2019, 51, 3915–3946. [Google Scholar]
- Ceunen, S.; Geuns, J.M.C. Steviol Glycosides: Chemical Diversity, Metabolism, and Function. J. Nat. Prod. 2013, 76, 1201–1228. [Google Scholar] [CrossRef]
- Moons, N.; De Borggraeve, W.; Dehaen, W. Stevioside and Steviol as Starting Materials in Organic Synthesis. Curr. Org. Chem. 2012, 16, 1986–1995. [Google Scholar] [CrossRef]
- Ukiya, M.; Sawada, S.; Kikuchi, T.; Kushi, Y.; Fukatsu, M.; Akihisa, T. Cytotoxic and Apoptosis-Inducing Activities of Steviol and Isosteviol Derivatives against Human Cancer Cell Lines. Chem. Biodivers. 2013, 10, 177–188. [Google Scholar] [CrossRef]
- Li, J.; Zhang, D.; Wu, X. Synthesis and biological evaluation of novel exo-methylene cyclopentanone tetracyclic diterpenoids as antitumor agents. Bioorg. Med. Chem. Lett. 2011, 21, 130–132. [Google Scholar] [CrossRef]
- Lin, Z.; Guo, Y.; Gao, Y.; Wang, S.; Wang, X.; Xie, Z.; Niu, H.; Chang, W.; Liu, L.; Yuan, H.; et al. ent-Kaurane Diterpenoids from Chinese Liverworts and Their Antitumor Activities through Michael Addition As Detected in Situ by a Fluorescence Probe. J. Med. Chem. 2015, 58, 3944–3956. [Google Scholar] [CrossRef]
- Zou, M.; Yu, S.-S.; Wang, K.; Zhang, D.-Y.; Wu, X.-M.; Hua, W.-Y. Glycosylation of ent-kaurene derivatives and an evaluation of their cytotoxic activities. Chin. J. Nat. Med. 2013, 11, 289–295. [Google Scholar] [CrossRef]
- Pezzuto, J.M.; Dhammika Nanayakkara, N.P.; Compadre, C.M.; Swanson, S.M.; Kinghorn, A.D.; Guenthner, T.M.; Sparnins, V.L.; Lam, L.K.T. Characterization of bacterial mutagenicity mediated by 13-hydroxy-ent-kaurenoic acid (steviol) and several structurally-related derivatives and evaluation of potential to induce glutathione S-transferase in mice. Mutat. Res. Toxicol. 1986, 169, 93–103. [Google Scholar] [CrossRef]
- Lin, S.-J.; Su, T.-C.; Chu, C.-N.; Chang, Y.-C.; Yang, L.-M.; Kuo, Y.-C.; Huang, T.-J. Synthesis of C-4-Substituted Steviol Derivatives and Their Inhibitory Effects against Hepatitis B Virus. J. Nat. Prod. 2016, 79, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Avent, A.G.; Hanson, J.R.; Hitchcock, P.B.; De Oliveira, B.H. The influence of a 15-hydroxy group on the rearrangement reactions of steviol and its 16,17-epoxide. J. Chem. Soc. Perkin Trans. 1 1990, 2661–2665. [Google Scholar] [CrossRef]
- BeMiller, J.N. Carbohydrate and Noncarbohydrate Sweeteners. In Carbohydrate Chemistry for Food Scientists; Elsevier: Duxford, UK, 2019; pp. 371–399. [Google Scholar]
- Wang, M.; Li, H.; Xu, F.; Gao, X.; Li, J.; Xu, S.; Zhang, D.; Wu, X.; Xu, J.; Hua, H.; et al. Diterpenoid lead stevioside and its hydrolysis products steviol and isosteviol: Biological activity and structural modification. Eur. J. Med. Chem. 2018, 156, 885–906. [Google Scholar] [CrossRef]
- Shi, L.-Y.; Wu, J.-Q.; Zhang, D.-Y.; Wu, Y.-C.; Hua, W.-Y.; Wu, X.-M. Efficient Synthesis of Novel Jolkinolides and Related Derivatives Starting from Stevioside. Synthesis 2011, 2011, 3807–3814. [Google Scholar]
- Mori, K.; Nakahara, Y.; Matsui, M. Diterpenoid total synthesis—XIX. Tetrahedron 1972, 28, 3217–3226. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Yu Fang, G. Highly regioselective and diastereoselective epoxidation of allylic amines with Oxone. Chem. Commun. 2005, 3448. [Google Scholar] [CrossRef]
- Kiss, L.; Forró, E.; Sillanpää, R.; Fülöp, F. Diastereo- and Enantioselective Synthesis of Orthogonally Protected 2,4-Diaminocyclopentanecarboxylates: A Flip from β-Amino- to β,γ-Diaminocarboxylates. J. Org. Chem. 2007, 72, 8786–8790. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Fülöp, F. Carbocyclic nucleosides from enantiomeric, α-pinane-based aminodiols. Tetrahedron Asymmetry 2010, 21, 831–836. [Google Scholar] [CrossRef]
- Le, T.M.; Csámpai, A.; Fülöp, F.; Szakonyi, Z. Regio- and Stereoselective Synthesis of Bicyclic Limonene-Based Chiral Aminodiols and Spirooxazolidines. Chem. Eur. J. 2018, 24, 13607–13615. [Google Scholar] [CrossRef]
- Le, T.M.; Szilasi, T.; Volford, B.; Szekeres, A.; Fülöp, F.; Szakonyi, Z. Stereoselective Synthesis and Investigation of Isopulegol-Based Chiral Ligands. Int. J. Mol. Sci. 2019, 20, 4050. [Google Scholar] [CrossRef] [Green Version]
- Jimeno, C.; Pastó, M.; Riera, A.; Pericàs, M.A. Modular Amino Alcohol Ligands Containing Bulky Alkyl Groups as Chiral Controllers for Et 2 Zn Addition to Aldehydes: Illustration of a Design Principle. J. Org. Chem. 2003, 68, 3130–3138. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | R1 | R2 | Yield (%) |
|---|---|---|---|---|
| 1 | 11 | H | Benzyl | 67 |
| 2 | 12 | Me | Benzyl | 75 |
| 3 | 13 | H | (R)-α-Methylbenzyl | 52 |
| 4 | 14 | H | (S)-α-Methylbenzyl | 59 |
| 5 | 15 | H | i-Pr | 57 |
| 6 | 16 | Et | Et | 63 |
| 7 | 17 | H | Propargyl | 72 |
| 8 | 18 | H | 3,5-bis(trifluoromethyl)benzyl | 32 |
| Entry | Ligand | Yield a (%) | eeb (%) | Major Configuration c |
|---|---|---|---|---|
| 1 | 11 | 78 | 23 | (R) |
| 2 | 12 | 85 | 52 | (R) |
| 3 | 17 | 77 | 33 | (R) |
| 4 | 18 | 85 | 31 | (R) |
| 5 | 19 | 79 | 30 | (R) |
| Compound | Conc (μM) | Growth Inhibition (%) ± SEM Calculated IC50 (μM) | |||
|---|---|---|---|---|---|
| A2780 | HeLa | SiHa | MDA-MB-231 | ||
| 5 | 10 | <20 * | <20 | <20 | <20 |
| 30 | <20 | <20 | 26.81 ± 2.02 | <20 | |
| 6 | 10 | <20 | <20 | <20 | <20 |
| 30 | <20 | <20 | <20 | <20 | |
| 7 | 10 | <20 | 27.07 ± 1.12 | <20 | <20 |
| 30 | 38.38 ± 2.53 | 34.72 ± 0.32 | <20 | <20 | |
| 8 | 10 | 49.78 ± 1.28 | 43.19 ± 2.20 | 96.41 ± 0.41 | <20 |
| 30 | 85.21 ± 0.53 | 51.98 ± 2.65 | 96.55 ± 0.30 | 52.04 ± 0.85 | |
| 10.18 | 21.89 | 4.64 | 29.90 | ||
| 9 | 10 | 22.24 ± 1.36 | <20 | <20 | <20 |
| 30 | 49.35 ± 0.58 | 26.80 ± 0.62 | 28.85 ± 0.69 | 34.62 ± 3.14 | |
| 10 | 10 | <20 | 25.61 ± 3.14 | <20 | <20 |
| 30 | 48.15 ± 0.68 | 28.46 ± 1.98 | <20 | 36.30 ± 3.34 | |
| 11 | 10 | 54.36 ± 3.34 | 37.41 ± 0.57 | <20 | <20 |
| 30 | 99.24 ± 0.16 | 98.58 ± 0.17 | 96.42 ± 0.44 | 98.45 ± 0.06 | |
| 6.68 | 9.37 | 24.68 | 26.16 | ||
| 12 | 10 | 35.57 ± 2.31 | 39.90 ± 2.76 | <20 | <20 |
| 30 | 65.79 ± 3.17 | 52.72 ± 2.04 | <20 | 23.77 ± 1.59 | |
| 17.34 | 23.49 | ||||
| 13 | 10 | 84.73 ± 0.84 | 60.46 ± 1.65 | 92.54 ± 0.77 | 94.09 ± 0.59 |
| 30 | 98.75 ± 0.17 | 98.81 ± 0.10 | 96.89 ± 0.93 | 98.30 ± 0.24 | |
| 4.19 | 4.79 | 6.07 | 4.32 | ||
| 14 | 10 | 89.99 ± 1.16 | 92.25 ± 0.99 | 91.23 ± 0.89 | 97.00 ± 0.16 |
| 30 | 98.87 ± 0.19 | 98.59 ± 0.07 | 94.34 ± 0.62 | 98.27 ± 0.20 | |
| 4.91 | 3.96 | 6.54 | 4.39 | ||
| 15 | 10 | <20 | <20 | <20 | <20 |
| 30 | <20 | 39.18 ± 1.84 | 20.41 ± 2.30 | <20 | |
| 16 | 10 | <20 | <20 | <20 | <20 |
| 30 | 29.07 ± 1.42 | 27.44 ± 1.06 | 20.83 ± 2.29 | <20 | |
| 17 | 10 | 20.75 ± 0.77 | <20 | <20 | <20 |
| 30 | 43.94 ± 2.99 | 30.21 ± 0.96 | <20 | 26.32 ± 1.04 | |
| 18 | 10 | 72.12 ± 1.13 | 69.15 ± 2.86 | 59.69 ± 1.52 | 97.68 ± 0.13 |
| 30 | 99.14 ± 0.12 | 98.25 ± 0.22 | 91.88 ± 1.34 | 98.26 ± 0.23 | |
| 6.25 | 5.73 | 7.84 | 4.76 | ||
| 19 | 10 | 98.53 ± 0.16 | 98.72 ± 0.11 | 96.50 ± 0.32 | 97.56 ± 0.42 |
| 30 | 98.97 ± 0.09 | 98.87 ± 0.03 | 97.06 ± 0.31 | 98.48 ± 0.40 | |
| 1.07 | 1.05 | 1.62 | 1.25 | ||
| 20 | 10 | 47.33 ± 0.91 | 42.91 ± 1.19 | <20 | <20 |
| 30 | 98.83 ± 0.23 | 98.95 ± 0.22 | 94.82 ± 0.05 | 95.81 ± 0.24 | |
| 9.78 | 10.39 | 14.95 | 15.09 | ||
| 21 | 10 | <20 | <20 | <20 | <20 |
| 30 | <20 | <20 | <20 | <20 | |
| 22 | 10 | 52.78 ± 2.29 | 56.41 ± 0.96 | 86.27 ± 1.83 | 83.98 ± 0.41 |
| 30 | 99.08 ± 0.06 | 99.01 ± 0.72 | 90.88 ± 1.03 | 98.09 ± 0.13 | |
| 8.60 | 4.13 | 8.58 | 6.58 | ||
| 23 | 10 | <20 | 23.98 ± 2.06 | 20.95 ± 1.64 | <20 |
| 30 | <20 | 44.56 ± 1.21 | 28.13 ± 0.75 | 89.42 ± 1.00 | |
| 24 | 10 | <20 | 20.53 ± 0.36 | <20 | <20 |
| 30 | 22.70 ± 0.56 | 28.92 ± 0.53 | <20 | <20 | |
| Cisplatin | 10 | 83.57 ± 1.21 | 42.61 ± 2.33 | 88.64 ± 0.50 | 67.51 ± 1.01 |
| 30 | 95.02 ± 0.28 | 99.93 ± 0.26 | 90.18 ± 1.78 | 87.75 ± 1.10 | |
| 1.30 | 12.43 | 7.84 | 3.74 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozsvár, D.; Nagy, V.; Zupkó, I.; Szakonyi, Z. Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpen Aminodiols. Int. J. Mol. Sci. 2020, 21, 184. https://doi.org/10.3390/ijms21010184
Ozsvár D, Nagy V, Zupkó I, Szakonyi Z. Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpen Aminodiols. International Journal of Molecular Sciences. 2020; 21(1):184. https://doi.org/10.3390/ijms21010184
Chicago/Turabian StyleOzsvár, Dániel, Viktória Nagy, István Zupkó, and Zsolt Szakonyi. 2020. "Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpen Aminodiols" International Journal of Molecular Sciences 21, no. 1: 184. https://doi.org/10.3390/ijms21010184
APA StyleOzsvár, D., Nagy, V., Zupkó, I., & Szakonyi, Z. (2020). Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpen Aminodiols. International Journal of Molecular Sciences, 21(1), 184. https://doi.org/10.3390/ijms21010184