Pathogenomics of Uterine Fibroids Development

{kind=link}
Abstract
:1. Introduction
2. The Origin of Fibroids
3. Tumor Initiation
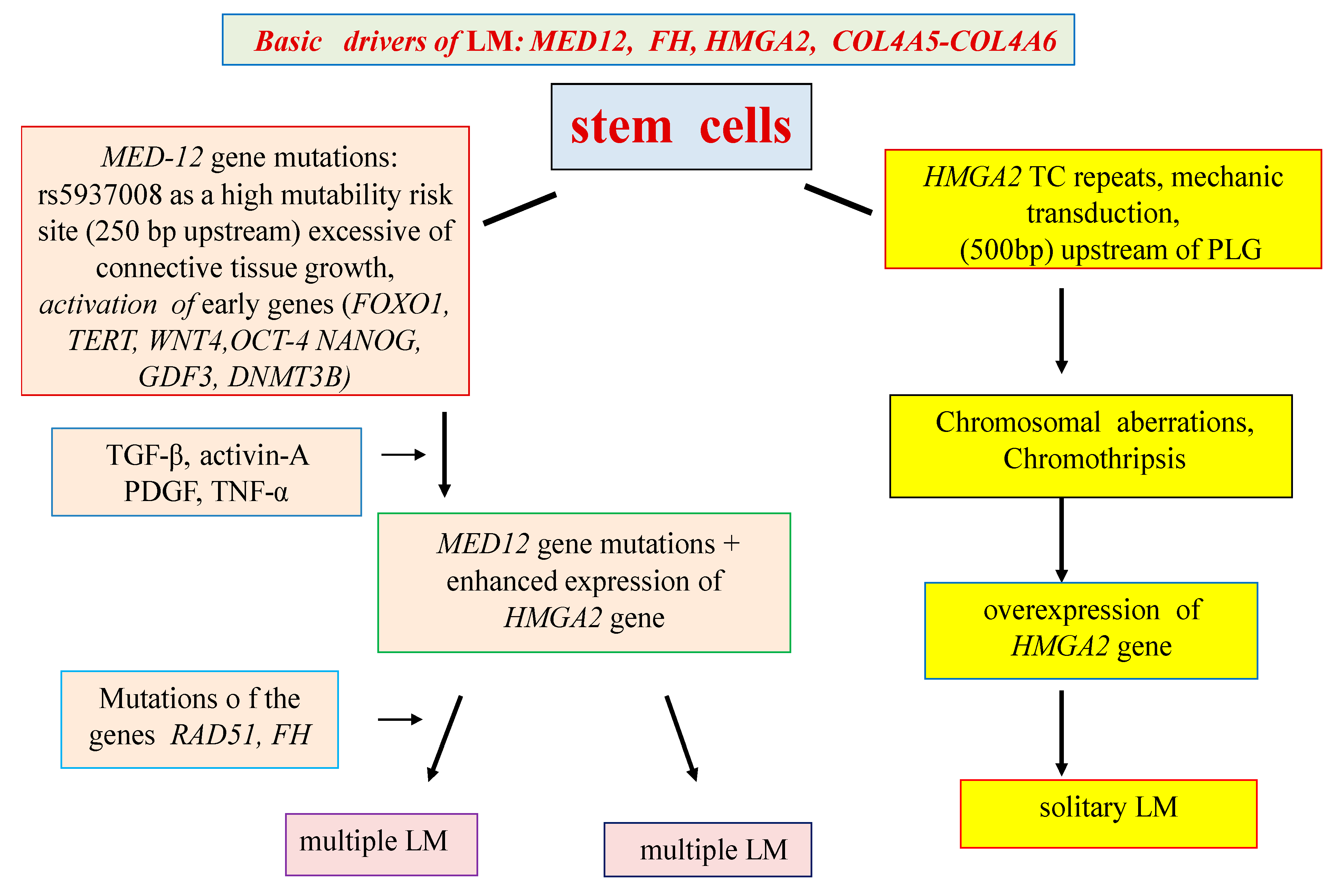
4. Genes Associated with the Development of LM
5. Epigenetic Regulation
6. General Considerations
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations:
| GWAS | genome-wide association studies |
| LM | leiomyoma |
| BrdU | 5-bromo-2–deoxyuridine |
| SC | stem cells |
| ECM | extracellular matrix |
| ERα | estrogen receptor α |
| PR | progesterone |
| FH | fumaratehydratase gene |
| TCF | transcription factor |
| TGF-β | transforming growth factor β |
| MAPK | mitogen-activated protein kinase |
| IGF | insulin-like growth factor |
| EGF | epidermal growth factor |
| HB-EGF | heparin-binding epidermal growth factor |
| CGH | comparative genomic hybridization |
References
- Bulun, S.E. Uterine Fibroids. N. Engl. J. Med. 2013, 369, 1344–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezhenar, V.F.; Komlichenko, E.V.; Yarmolinskaya, M.I.; Dedul, A.G.; Sheveleva, T.S.; Malushko, A.V.; Kalinina, E.A.; Zubareva, T.M.; Gamzatova, Z.K.; Kondratyev, A.A. Innovative approaches to reproductive function recovery in patients with uterine myoma. Akusherstvo iginekologiya/Obstet. Gynecol. 2016, 1. [Google Scholar] [CrossRef]
- Tonoyan, N.M.; Kozachenko, I.F.; Frankevich, V.E.; Chagovets, V.V.; Adamyan, L.V. Recurrences of uterine fibroids. The modern view on the problems of diagnosis, treatment, and prognosis. Obstet. Gynegology 2019, 3. [Google Scholar] [CrossRef]
- Soghoyan, N.S.; Adamyan, L.V. Genetic mechanisms of uterine leiomyoma. Probl. Reprod. 2016, 22, 22–34. [Google Scholar] [CrossRef]
- Baranov, V.S.; Ivaschenko, T.E.; Yarmolinskaya, M.I. Comparative systems genetics view of endometriosis and uterine leiomyoma: Two sides of the same coin? Syst. Biol. Reprod. Med. 2016, 62, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Canevari, R.A.; Pontes, A.; Rosa, F.E.; Rainho, C.A.; Rogatto, S.R. Independent clonal origin of multiple uterine leiomyomas that was determined by X chromosome inactivation and microsatellite analysis. Am. J. Obs. Gynecol. 2005, 193, 1395–1403. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, C.; Hao, J.; Sung, C.J.; Quddus, M.R.; Steinhoff, M.M. Use of X-chromosome inactivation pattern to determine the clonal origins of uterine leiomyoma and leiomyosarcoma. Hum Pathol. 2006, 37, 1350–1356. [Google Scholar] [CrossRef]
- Ono, M.; Maruyama, T.; Masuda, H.; Kajitani, T.; Nagashima, T.; Arase, T.; Ito, M.; Ohta, K.; Uchida, H.; Asada, H.; et al. Side population in human uterine myometrium displays phenotypic and functional characteristics of myometrial stem cells. Proc Natl Acad Sci USA. 2007, 104, 18700–18705. [Google Scholar] [CrossRef] [Green Version]
- Mas, A.; Cervello, I.; Gil-Sanchis, C.; Simon, C. Current understanding of somatic stem cells in leiomyoma formation. Fertil. Steril. 2014, 102, 613–620. [Google Scholar] [CrossRef]
- Ono, M.; Bulun, S.E.; Maruyama, T. Tissue-Specific Stem Cells in the Myometrium and Tumor-Initiating Cells in Leiomyoma. Biol. Reprod. 2014, 91, 1–7. [Google Scholar] [CrossRef]
- Ciavattini, A.; Giuseppe, D.J.; Storton, P.; Montik, N.; Giannubilo, S.R.; Litta, P.; Islam, S.; Tranquilli, A.L.; Reis, F.M.; Ciarmela, P. Uterine Fibroids: Pathogenesis and Interactions with Endometrium and Endomyometrial Junction. Obstet Gynecol Int. 2013, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Kajitani, T.; Uchida, H.; Arase, T.; Oda, H.; Uchida, S.; Ota, K.; Nagashima, T.; Masuda, H.; Miyazaki, K.; et al. CD34 and CD49f Double-Positive and Lineage Marker-Negative Cells Isolated from Human Myometrium Exhibit Stem Cell-Like Properties Involved in Pregnancy-Induced Uterine Remodeling. Biol. Reprod. 2015, 93, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, P.; Ono, M.; Moravek, M.B.; John, S.; Coon, V.; Navarro, A.; Monsivais, D.; Dyson, M.T.; Druschitz, S.A.; Malpani, S.S.; et al. Human Uterine Leiomyoma Stem/Progenitor Cells Expressing CD34 and CD49b Initiate Tumors In Vivo. J. Clin. Endocrinol Metab. 2015, 100, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Brakta, S.; Mas, A.; Al-Hendy, A. The ontogeny of myometrial stem cells in OCT4-GFP transgenic mouse model. Stem. Cell Res. Ther. 2018, 9, 333. [Google Scholar] [CrossRef] [PubMed]
- Moroni, R.M.; Vieira, C.S.; Ferriani, R.A.; Reis, R.M.; Nogueira, A.A.; Brito, L.G. Presentation and treatment of uterine leiomyoma in adolescence: A systematic review. BMC Womens Health. 2015, 15, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.S.; Ciavattini, A.; Petraglia, F.; Castellucci, M.; Ciarmela, P. Extracellular matrix in uterine leiomyoma pathogenesis: a potential target for future therapeutics. Hum Reprod Update 2018, 24, 59–85. [Google Scholar] [CrossRef] [Green Version]
- Thorne, J.T.; Segal, T.R.; Chang, S.; Jorge, S.; Segars, J.H.; Leppert, P.C. Dynamic Reciprocity Between Cells and Their Microenvironment in Reproduction. Biol Reprod. 2015, 92, 1–10. [Google Scholar] [CrossRef]
- Leppert, P.C.; Jayes, F.L.; Segars, J.H. The Extracellular Matrix Contributes to Mechanotransduction in Uterine Fibroids. Obstet Gynecol Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mehine, M.; Kaasinen, E.; Mäkinen, N.; Katainen, R.; Kämpjärvi, K.; Pitkänen, E.; Heinonen, H.R.; Bützow, R.; Kilpivaara, O.; Kuosmanen, A.; et al. Characterization of Uterine Leiomyomas by Whole-Genome Sequencing. N. Engl. J. Med. 2013, 369, 43–53. [Google Scholar] [CrossRef]
- Medikare, V.; Kandukuri, L.R.; Ananthapur, V.; Deenadayal, M.; Nallari, P. The Genetic Bases of Uterine Fibroids; A Review. Reprod Infertil. 2011, 12, 181–191. [Google Scholar]
- Pendina, A.A.; Koltsova, A.S.; Efimova, O.A.; Malysheva, O.V.; Osinovskaya, N.S.; Sultanov, I.Y.; Tikhonov, A.V.; Shved, N.Y.; Chiryaeva, O.G.; Simareva, A.D.; et al. Case of chromothripsis in a large solitary non-recurrent uterine leiomyoma. Eur. J. Obstet. Gynecol. Reprod. Biol. 2017, 219, 134–136. [Google Scholar] [CrossRef] [PubMed]
- Koltsova, A.S.; Pendina, A.A.; Efimova, O.A.; Chiryaeva, O.G.; Kuznetzova, T.V.; Baranov, V.S. On the Complexity of Mechanisms and Consequences of Chromothripsis: An Update. Front Genet. 2019, 10, 393. [Google Scholar] [CrossRef] [PubMed]
- Lynch, A.M.; Morton, C.C. Uterus: Leiomyoma. Atlas Genet Cytogenet Oncol Haematol. 2008, 12, 68–73. [Google Scholar] [CrossRef]
- Bowden, W.; Skorupski, J.; Kovanci, E.; Rajkovic, A. Detection of novel copy numbervariants in uterine leiomyomas usinghigh-resolution SNP arrays. Mol. Hum Reprod. 2009, 15, 563–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehine, M.; Mäkinen, N.; Heinonen, H.R.; Aaltonen, L.A.; Vahteristo, P. Genomics of uterine leiomyomas: insights from high-throughput sequencing. Fertil. Steril. 2014, 102, 621–629. [Google Scholar] [CrossRef]
- Makinen, N.; Mehine, M.; Tolvanen, J.; Kaasinen, E.; Li, Y.; Lehtonen, H.J.; Gentile, M.; Yan, J.; Enge, M.; Taipale, M.; et al. MED12, the Mediator Complex Subunit 12 Gene, Is Mutated at High Frequency in Uterine Leiomyomas. Science 2011, 334, 252–255. [Google Scholar] [CrossRef]
- Osinovskaya, N.S.; Malysheva, O.V.; Shved, N.Y.; Ivashchenko, T.E.; Sultanov, I.Y.; Efimova, O.A.; Yarmolinskaya, M.I.; Bezhenar, V.F.; Baranov, V.S. Frequency and Spectrum of MED12 Exon 2 Mutations in Multiple Versus Solitary Uterine Leiomyomas From Russian Patients. Int. J. Gynecol Pathol. 2016, 35, 509–515. [Google Scholar] [CrossRef]
- Heikkinen, T.; Äyräväinen, A.; Hänninen, J.; Ahvenainen, T.; Bützow, R.; Pasanen, A.; Vahteristo, P. MED12 mutations and fumarate hydratase inactivation in uterine adenomyomas. Hum. Reprod Open 2018, 4, 1–8. [Google Scholar] [CrossRef]
- Cui, T.; Leng, F. Specific Recognition of AT-Rich DNA Sequences by the Mammalian HighMobility Group Protein AT-hook 2: A SELEX Study. Biochemistry 2007, 46, 13059–13066. [Google Scholar] [CrossRef]
- Borrmann, L.; Seebeck, B.; Rogalla, P.; Bullerdiek, J. Human HMGA2 promoter is coregulated by a polymorphic dinucleotide (TC)-repeat. Oncogene 2003, 22, 756–760. [Google Scholar] [CrossRef] [Green Version]
- Lehtonen, R.; Kiuru, M.; Vanharanta, S.; Sjoberg, J.; Aaltonen, L.M.; Aittomaki, K.; Arola, J.; Butzow, R.; Eng, C.; Husgafvel-Pursiainen, K.; et al. Biallelic Inactivation of Fumarate Hydratase (FH) Occurs in Nonsyndromic Uterine Leiomyomas but Is Rare in Other Tumors. Am. J. pathol. 2004, 164, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Mehine, M.; Kaasinen, E.; Heinonen, H.R.; Mäkinen, N.; Kämpjärvi, K.; Sarvilinna, N.; Aavikko, M.; Vähärautio, A.; Pasanen, A.; Bützow, R.; et al. Integrated data analysis reveals uterine leiomyoma subtypes with distinct driver pathways and biomarkers. Proc. Natl. Acad Sci. USA 2016, 113, 1315–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertsch, E.; Qiang, W.; Zhang, Q.; Espona-Fiedler, M.; Druschitz, S.; Liu, Y.; Mittal, K.; Kong, B.; Kurita, T.; Wei, J.J. MED12 and HMGA2 mutations: Two independent genetic events in uterine leiomyoma and leiomyosarcoma. Mod. Pathol. 2014, 27, 8–1144. [Google Scholar] [CrossRef] [PubMed]
- Galindo, L.J.; Hernández-Beeftink, T.; Salas, A.; Jung, Y.; Reyes, R.; de Oca, F.M.; Hernández, M.; Almeida, T.A. HMGA2 and MED12 alterations frequently co-occur in uterine leiomyomas. Gynecol Oncol. 2018, 150, 562–568. [Google Scholar] [CrossRef]
- Välimäki, N.; Kuisma, H.; Pasanen, A.; Heikinheimo, O.; Sjöberg, J.; Bützow, R.; Sarvilinna, N.; Heinonen, H.R.; Tolvanen, J.; Bramante, S.; et al. Genetic predisposition to uterine leiomyoma is determined by loci for genitourinary development and genome stability. Elife 2018, 7, 50. [Google Scholar] [CrossRef]
- Xie, J.; Ubango, J.; Ban, Y.; Chakravarti, D.; Kim, J.J.; Wei, J.J. Comparative analysis of AKT and the related biomarkers in uterine leiomyomas with MED12, HMGA2, and FH mutations. Genes Chromosomes Cancer 2018, 57, 485–494. [Google Scholar] [CrossRef]
- Dzhemlikhanova, L.K.; Efimova, O.A.; Osinovskaya, N.S.; Parfenyev, S.E.; Niauri, D.A.; Sultanov, I.Y.; Malysheva, O.V.; Pendina, A.A.; Shved, N.Y.; Ivashchenko, T.E. Catechol-O-methyltransferase Val158Met polymorphism is associated with increased risk of multiple uterine leiomyomas either positive or negative for MED12 exon 2 mutations. J. Clin. Pathol. 2017, 70, 233–236. [Google Scholar] [CrossRef]
- Osinovskaya, N.S.; Ivashchenko, T.E.; Dolinskii, A.K.; Sultanov, I.Y.; Ghimbovschi, S.; Hoffman, E.; Bezhenar’, V.F.; Baranov, V.S. MED12 gene mutations in women with uterine myoma. Russ. J. Genet. 2013, 49, 1245–1249. [Google Scholar] [CrossRef]
- Kim, J.J.; Sefton, E.C. The role of progesterone signaling in the pathogenesis of uterine leiomyoma. Mol Cell Endocrinol. 2012, 358, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, F.; Pabalan, N.; Ekaratcharoenchai, N.; Neto, A.S.; Christofolini, D.M.; de Oliveira, R.; Bianco, B.; Barbosa, C.P. PROGINS Polymorphism of the Progesterone Receptor Gene and the Susceptibility to Uterine Leiomyomas: A Systematic Review and Meta-Analysis. Genet Test Mol. Biomark. 2018, 22, 295–301. [Google Scholar] [CrossRef]
- Benassayag, C.; Leroy, M.J.; Rigourd, V.; Robert, B.; Honoré, J.C.; Mignot, T.M.; Vacher-Lavenu, M.C.; Chapron, C.; Ferré, F. Estrogen receptors (ERa/ERb) in normal and pathological growth of the human myometrium: pregnancy and leiomyoma. Am. J. Physiol. 1999, 276, 1112–1118. [Google Scholar] [CrossRef]
- Lora, V.; Grings, A.O.; Capp, E.; von Eye Corleta, H.; Brum, I.S. Gene and protein expression of progesterone receptor isoforms A and B, p53 and p21 in myometrium and uterine leiomyoma. Arch Gynecol Obstet. 2012, 286, 119–124. [Google Scholar] [CrossRef]
- Tal, R.; Segars, J.H. The role of angiogenic factors in fibroid pathogenesis: potential implicationsfor future therapy. Hum Reprod Update 2014, 20, 194–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norian, J.M.; Malik, M.; Parker, C.Y.; Joseph, D.; Leppert, P.C.; Segars, J.H.; Catherino, W.H. Transforming Growth Factor β3 Regulates the Versican Variants in the Extracellular Matrix-Rich Uterine Leiomyomas. Reprod Sci. 2009, 16, 1153–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, M.; Segars, J.; Catherino, W.H. Why Leiomyomas Are Called Fibroids: The Central Role of Extracellular Matrix in Symptomatic Women. Matrix Biol. 2012, 31, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Feofilova, M.A.; Tomareva, E.I.; Evdokimova, D.V. Etiology And Pathogenesis Of Uterine Myoma, Its Relationship With Health And Reproductive Function Of Women (Literature Review). J. New Med. Technol. 2017, 24, 249–260. [Google Scholar] [CrossRef]
- Malik, M.; Norian, J.; McCarthy-Keith, D.; Britten, J.; Catherino, W. Why Leiomyomas Are Called Fibroids: The Central Role of Extracellular Matrix in Symptomatic Women. Semin. Reprod. Med. 2010, 28, 169–179. [Google Scholar] [CrossRef]
- Gloria-Bottini, F.; Ammendola, M.; Saccucci, P.; Pietropolli, A.; Neri, A.; Magrini, A. Allergy and Uterine Leiomyomas: Cooperative Interaction with ACP1 Genetic Polymorphism. Egidio BottiniJ Reprod Infertil. 2015, 16, 199–202. [Google Scholar]
- Navarro, A.; Yin, P.; Ono, M.; Monsivais, D.; Moravek, M.B.; Coon, J.S.; Dyson, M.T.; Wei, J.-J.; Bulun, S.E. 5-Hydroxymethylcytosine Promotes Proliferation of Human Uterine Leiomyoma: A Biological Link to a New Epigenetic Modification in Benign Tumors. J. Clin. Endocrinol Metab. 2014, 99, 2437–2445. [Google Scholar] [CrossRef]
- Liu, S.; Yin, P.; Kujawa, S.A.; Coon, J.S.; Okeigwe, I.; Bulun, S.E. Progesterone receptor integrates the effects of mutated MED12 and altered DNA methylation to stimulate RANKL expression and stem cell proliferation in uterine leiomyoma. Oncogene 2019, 38, 2722–2735. [Google Scholar] [CrossRef] [Green Version]
- Kol’tsova, A.S.; Pendina, A.A.; Efimova, O.A.; Kaminskaya, A.N.; Tikhonov, A.V.; Osinovskaya, N.S.; Sultanov, I.Y.; Shved, N.Y.; Kakhiani, M.I.; Baranov, V.S. Differential DNA Hydroxymethylation in Human Uterine Leiomyoma Cells Depending on the Phase of Menstrual Cycle and Presence of MED12 Gene Mutations. Bull Exp Biol Med. 2017, 163, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, Y.; Maekawa, R.; Asada, H.; Taketani, T.; Tamura, I.; Tamura, H.; Ogane, J.; Hattori, N.; Shiota, K.; Sugino, N. Aberrant DNA methylation status in human uterine leiomyoma. Mol Hum Reprod. 2009, 15, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Zhang, X.; Obijuru, L.; Laser, J.; Aris, V.; Lee, P.; Mittal, K.; Soteropoulos, P.; Wei, J.J. A Micro-RNA Signature Associated with Race, Tumor Size, and Target Gene Activity in Human Uterine Leiomyomas. Genes Chromosomes Cancer 2007, 46, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Karmon, A.E.; Cardozo, E.R.; Rueda, B.R.; Styer, A.K. MicroRNAs in the development and pathobiology of uterine leiomyomata: does evidence support future strategies for clinical intervention? Hum Reprod Update. 2014, 20, 670–687. [Google Scholar] [CrossRef]
- Borahay, M.A.; Al-Hendy, A.; Kilic, G.S.; Boehning, D. Signaling Pathways in Leiomyoma: Understanding Pathobiology and Implications for Therapy. Mol. Med. 2015, 21, 242–256. [Google Scholar] [CrossRef]
- Rafnar, T.; Gunnarsson, B.; Stefansson, O.A.; Sulem, P.; Ingason, A.; Frigge, M.L.; Stefansdottir, L.; Sigurdsson, J.K.; Tragante, V.; Steinthorsdottir, V. Variants associating with uterine leiomyoma highlight genetic background shared by various cancers and hormone-related traits. Nat Commun. 2018, 9, 3636. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baranov, V.S.; Osinovskaya, N.S.; Yarmolinskaya, M.I. Pathogenomics of Uterine Fibroids Development. Int. J. Mol. Sci. 2019, 20, 6151. https://doi.org/10.3390/ijms20246151
Baranov VS, Osinovskaya NS, Yarmolinskaya MI. Pathogenomics of Uterine Fibroids Development. International Journal of Molecular Sciences. 2019; 20(24):6151. https://doi.org/10.3390/ijms20246151
Chicago/Turabian StyleBaranov, Vladislav S., Natalia S. Osinovskaya, and Maria I. Yarmolinskaya. 2019. "Pathogenomics of Uterine Fibroids Development" International Journal of Molecular Sciences 20, no. 24: 6151. https://doi.org/10.3390/ijms20246151
APA StyleBaranov, V. S., Osinovskaya, N. S., & Yarmolinskaya, M. I. (2019). Pathogenomics of Uterine Fibroids Development. International Journal of Molecular Sciences, 20(24), 6151. https://doi.org/10.3390/ijms20246151