3. Discussion
Due to the involvement of CK1 isoforms in numerous cellular processes, dysregulation of CK1 can be linked to the development of different cancers as well as to neurodegenerative diseases. Apart from an increase of expression levels, elevated CK1δ kinase activity can also be caused by mutations occurring in the genomic sequence coding for CK1δ. In order to pharmacologically control CK1δ activity, several small molecule inhibitors are already available. However, CK1 isoform-specificity and pharmacokinetic parameters for most of these inhibitors are not satisfactory and new inhibitor compounds with improved properties are required. Furthermore, specific targeting of CK1δ mutants occurring in certain tumor entities would provide a broad therapeutic window for the treatment of the respective tumor without inducing severe off-target effects in healthy tissue. In order to deal with these issues, in the present study we (i) characterized enzyme kinetic properties of CK1δ mutants identified in various cancer entities including the colon and rectum and (ii) we characterized the CK1δ wild type- and mutant-specific inhibitory potential of a small set of CK1δ-specific small molecule inhibitors in vitro and on four colon cancer cell lines.
So far, only few CK1δ mutants have been described in the literature, which could be linked to the pathogenesis of certain diseases. For instance, mutation CK1δ
S97C has been associated with the development of breast cancer and ductal carcinoma [
1]. Mutation CK1δ
R324H has been linked to increased oncogenic potential and was detected in a patient with large and multiple colonic polyps. In an experimental setting, CK1δ
R324H has been shown to potently transform RKO colon cancer cells [
3]. Although both mutants are not associated with an immediate increase of kinase activity, at least for CK1δ
R324H, a mechanism has been detected leading to Wnt/β-catenin-independent morphogenetic movements in early
Xenopus development [
3]. Furthermore, a hyperactive mutant (CK1δ
T67S) has been found in colon and rectal cancer and expression of CK1δ
T67S resulted in higher oncogenic potential in cell culture experiments as well as in a mouse xenotransplantation model [
2].
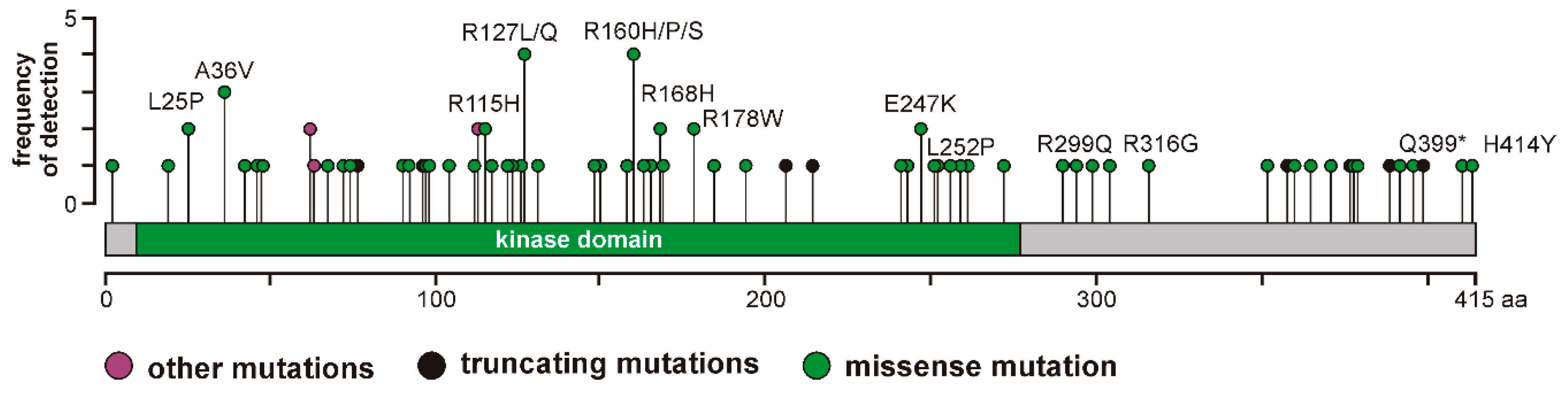
In addition, 123 mutations affecting the
CSNK1D gene are currently listed in the database cBioPortal for Cancer Genomics [
5,
6]. However, the effects of these mutants on CK1δ kinase activity and their oncogenic potential has not been analyzed so far. Therefore, we selected a panel of CK1δ mutations (L25P, A36V, R115H, R127L, R127Q, I148M, R160H, R160P, R160S, R168H, R178W, E247K, L252P, R299Q, R316G, Q399*, and H414Y) to subsequently perform analysis of enzyme kinetic parameters.
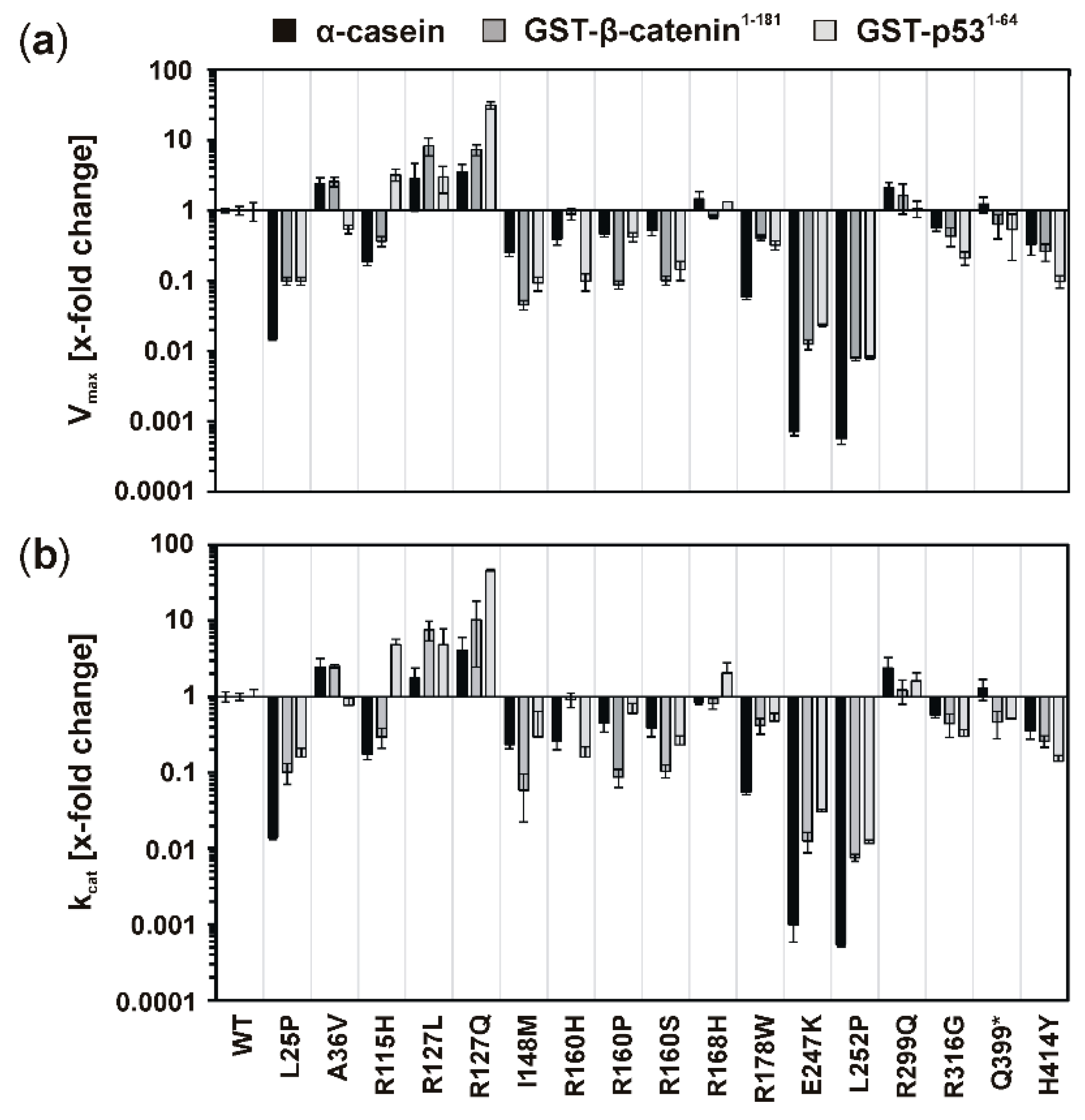
Elevated kinase activity could be determined for mutants GST-CK1δR127L and GST-CK1δR127Q, which can be explained by changes in the electrical charge of the protein resulting from the exchange of the positively charged amino acid arginine to hydrophobic leucine or polar, but uncharged, glutamine. These changes in protein charge might result in conformational changes affecting kinase activity. Moreover, the catalytically essential residue Arg-128 is located in direct proximity to the mutated position 127, and the entire sequence between amino acids 128–133 belongs to the catalytic loop of CK1δ. Therefore, mutations within this sequence or in close proximity to it could be linked to alteration of kinase activity.
Remarkably decreased kinase activity could be observed for GST-CK1δ mutants carrying mutations I148M, R160H, R160P, R160S, R168H, R178W, E247K, or L252P. Interestingly, for mutants CK1δ
E257K and CK1δ
L252P, nearly no residual kinase activity could be determined in enzyme kinetic analysis. Therefore, both mutants can be considered kinase-dead mutants. So far, also mutant CK1δ
K38M, bearing an amino acid change at residue Lys-38, which is involved in binding of ATP, has been described as kinase-dead mutant of CK1δ [
31,
32].
Mutations located in the C-terminus of CK1δ (ranging from amino acid 296 to 415 for human CK1δ transcription variant 1 [
33]) resulted in different effects on phosphorylation of α-casein in kinetic analysis. While increased activity was determined for GST-CK1δ
R299Q and GST-CK1δ
Q399*, decreased activity was observed for GST-CK1δ
R316G and GST-CK1δ
H414Y. In general, several C-terminal residues of CK1δ are involved in regulation of kinase activity, mostly by phosphorylation events mediated by intramolecular autophosphorylation or by phosphorylation by upstream kinases [
21,
34,
35,
36]. Consequently, if this domain is affected by conformational changes due to amino acid exchanges also regulation of kinase activity might be affected.
In general, mutations in the CK1δ protein might not only affect kinase activity, but can also result in different binding of small molecule inhibitors. This has previously been shown for the hyperactive mutant CK1δ
T67S as well as for different phosphorylation-site mutants [
2,
21]. Considering a potential use in therapeutic strategies for personalized medicine, stronger binding of inhibitor compounds to hyperactive CK1δ mutants could be of outstanding benefit. By selectively inhibiting CK1δ mutants expressed in tumor tissue, a wider therapeutic window is opened and side effects on tissues expressing wild type CK1δ can be limited or even avoided. In order to identify differences in sensitivity toward inhibition by specific small molecule inhibitors, certain inhibitor compounds were tested for their effects on CK1δ mutants. Compared to wild type CK1δ, the hyperactive GST-CK1δ
R127Q mutant could be stronger inhibited by all selected inhibitor compounds. Inhibitor-specific effects determined for incubation of GST-CK1δ
R127L and GST-CK1δ
R299Q with IWP-2 were also stronger than for wild type CK1δ and similar significant observations could as well be made for inhibition of GST-CK1δ
A36V and GST-CK1δ
R115H with Bischof-5. Mutant GST-CK1δ
R168H was significantly stronger inhibited by both Bischof-5 and Richter-2 when being compared to inhibitory effects observed on wild type CK1δ. In contrast, mutations in CK1δ can also lead to reduced inhibition by CK1δ-specific inhibitors. This could be mainly observed for Richter-2 and GST-CK1δ mutants A36V, R115H, and R127L, as well as for IWP-4 and mutants R299Q and Q399*.
Expression of hyperactive CK1δ mutants has been shown to be associated with increased oncogenic potential, while decreased activity of CK1δ results in reduced oncogenic functions [
2,
4]. Consequently, by treating tumor cells expressing hyperactive CK1δ mutants with CK1δ-specific inhibitors, beneficial effects could be obtained. With the purpose to screen for these beneficial effects, CK1δ mutant-expressing cells were treated with CK1δ-specific inhibitors. For being analyzed in this experimental approach, three CK1δ mutants were selected: mutant CK1δ
R127Q presented the highest activity among all tested mutants, and mutant CK1δ
R168H represented a unique hyperactive mutation located between Ile-148 and Leu-252. In addition, the previously described mutant CK1δ
T67S, being characterized by increased kinase activity and high oncogenic potential, was tested in the selected cell culture model.
In order to identify inhibitors selectively targeting wild type and/or mutant CK1 isoforms, two sets of newly developed inhibitor compounds were tested within the present study. These compounds were designed based on previously reported IWP-derivatives, which demonstrated high potency and selectivity for CK1δ as well as increased affinity to a CK1δ
M82F gatekeeper mutant [
15]. Initial screening of the IWP-derived compounds
16,
17,
18, and
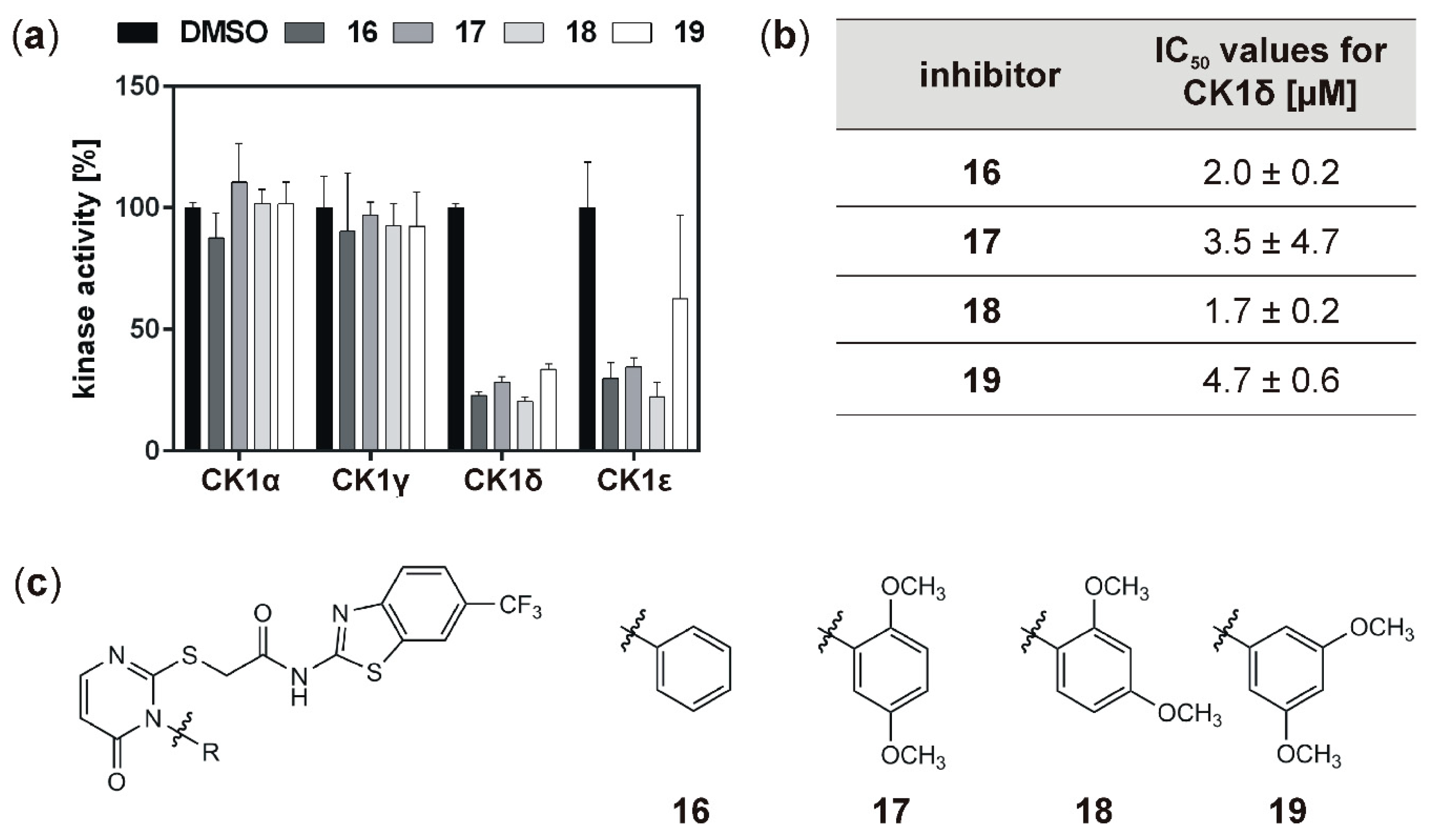
19 indicated that all compounds strongly inhibit CK1δ (and ε) with IC
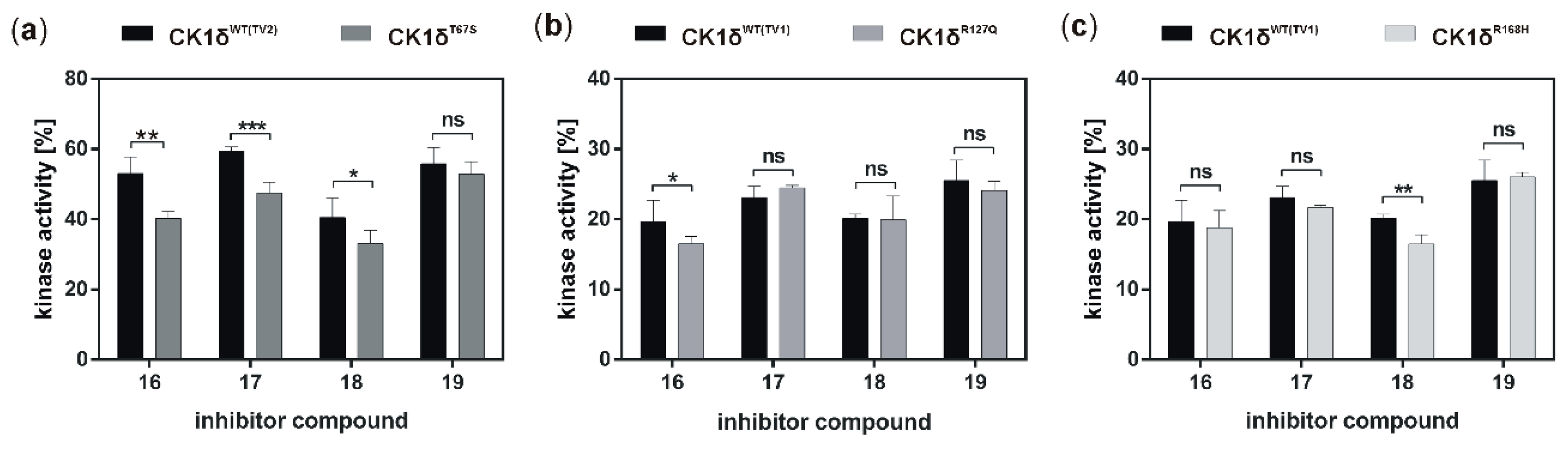
50 values within the one-digit micromolar range. While, in general, inhibition of the hyperactive CK1δ mutants T67S, R127Q, and R168H was within the same range as observed for wild type CK1δ, for few of the tested inhibitors slightly stronger inhibition of the mutants could be detected.
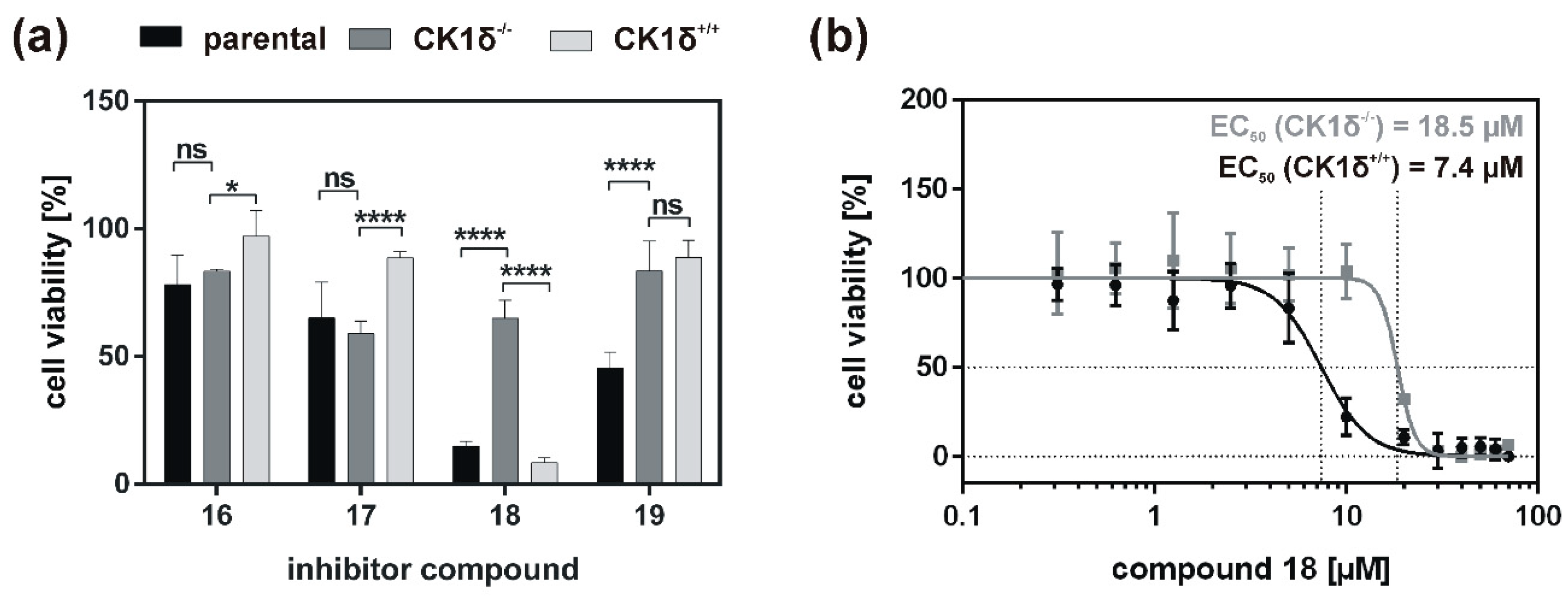
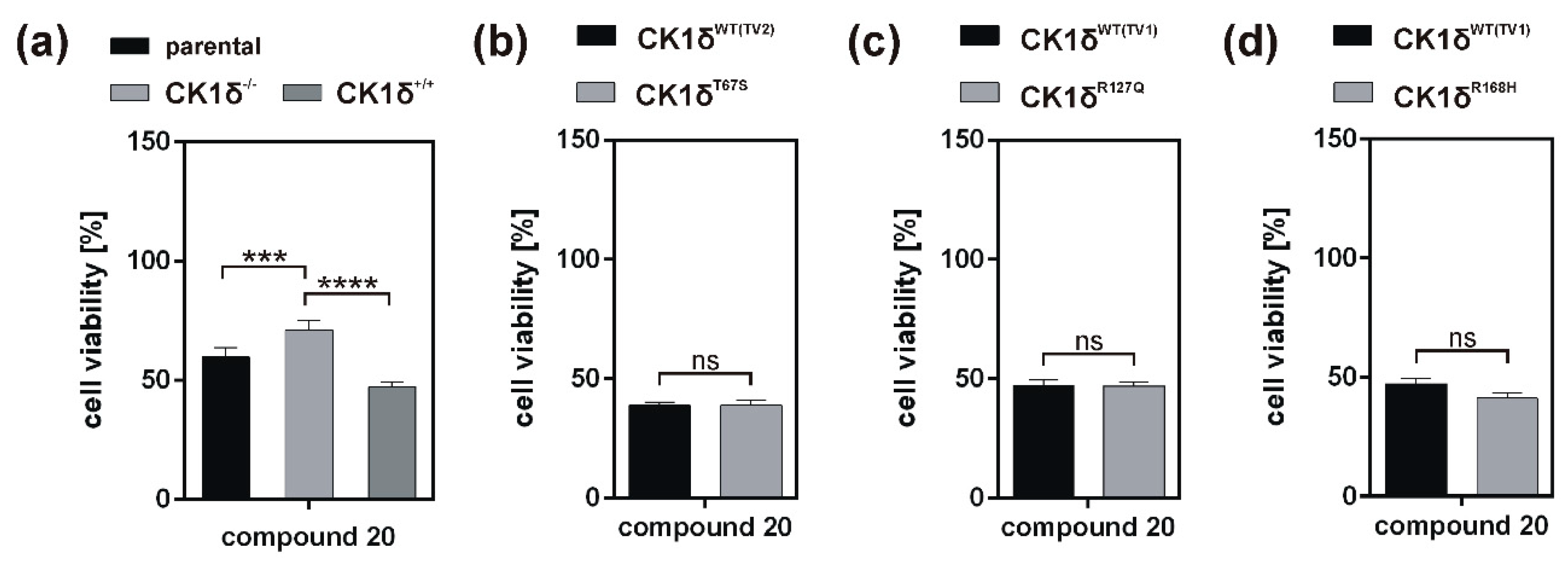
Analysis of inhibitory effects of the newly designed inhibitors on cultured cells was performed using HeLa cells. By also using HeLa cells, being deficient for CK1δ (HeLa CK1δ−/−), the target selectivity of the tested inhibitor compounds could be analyzed. Interestingly, treatment with compound 18 and to a lesser extent also with compound 19 (or compound 20, which was tested in a later stage of the study) showed stronger effects on CK1δ-expressing cells than on HeLa CK1δ−/− cells. We therefore conclude that at least compound 18 selectively targets CK1δ. This conclusion is supported by EC50 data generated for compound 18, showing a significantly increased EC50 value for HeLa CK1δ−/− cells. However, the effects observed for HeLa CK1δ−/− cells are still obvious, which might be caused by simultaneous inhibition of other CK1 isoforms like the highly related isoform CK1ε. Furthermore, the effects obtained for compounds 16 and 17 might result from off-target effects on other CK1 isoforms or other cellular kinases in general. Significantly decreased effects on cell viability of HeLa CK1δ+/+ cells in comparison to parental cells could be explained by overexpression of exogenous CK1δ in HeLa CK1δ+/+ cells, potentially leading to highly increased intracellular target concentration.
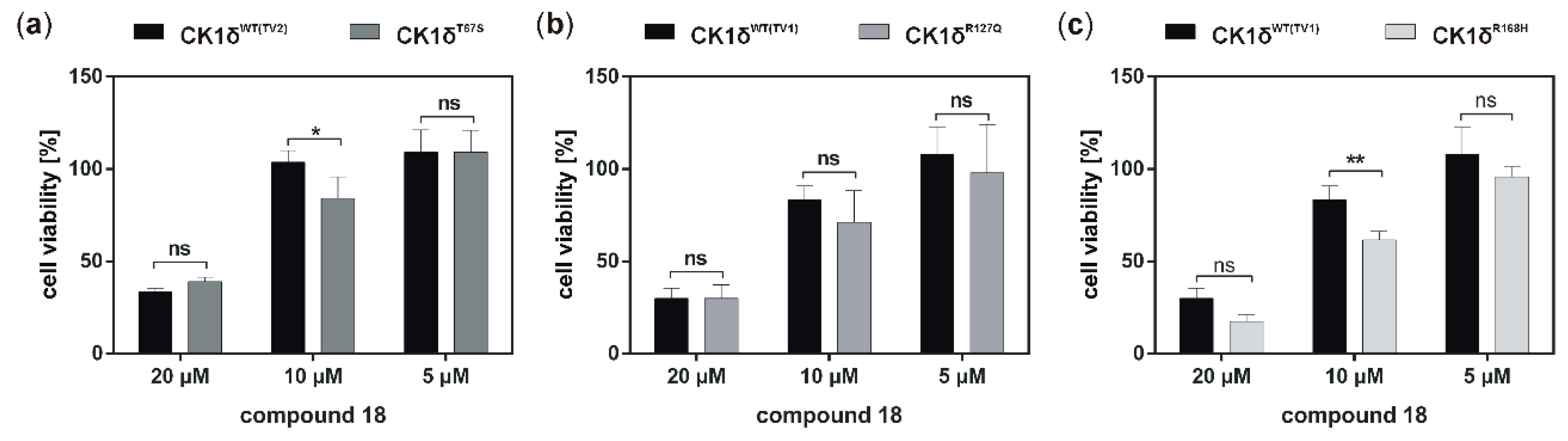
If the hyperactive CK1δ mutants T67S, R127Q, and R168H were expressed in HeLa CK1δ−/− cells, inhibitory effects mediated by compound 18 were slightly stronger. This finding underlines the marginally increased affinity of compound 18 to CK1δ mutants in comparison to wild type CK1δ. Interestingly, for all three mutant-expressing cell lines, dose-dependently different effects could be observed, thereby demonstrating the need for proper dose-response investigation.
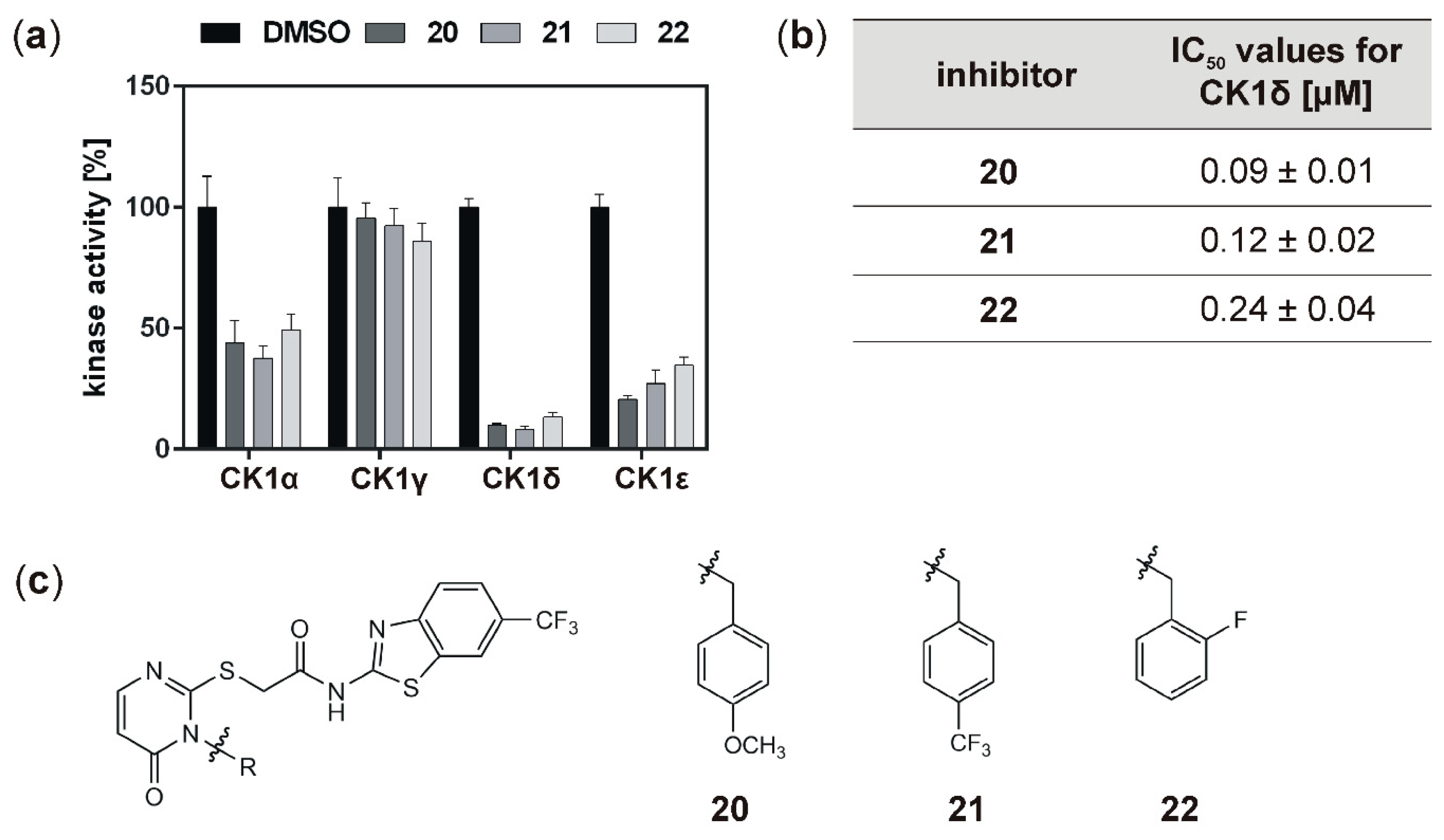
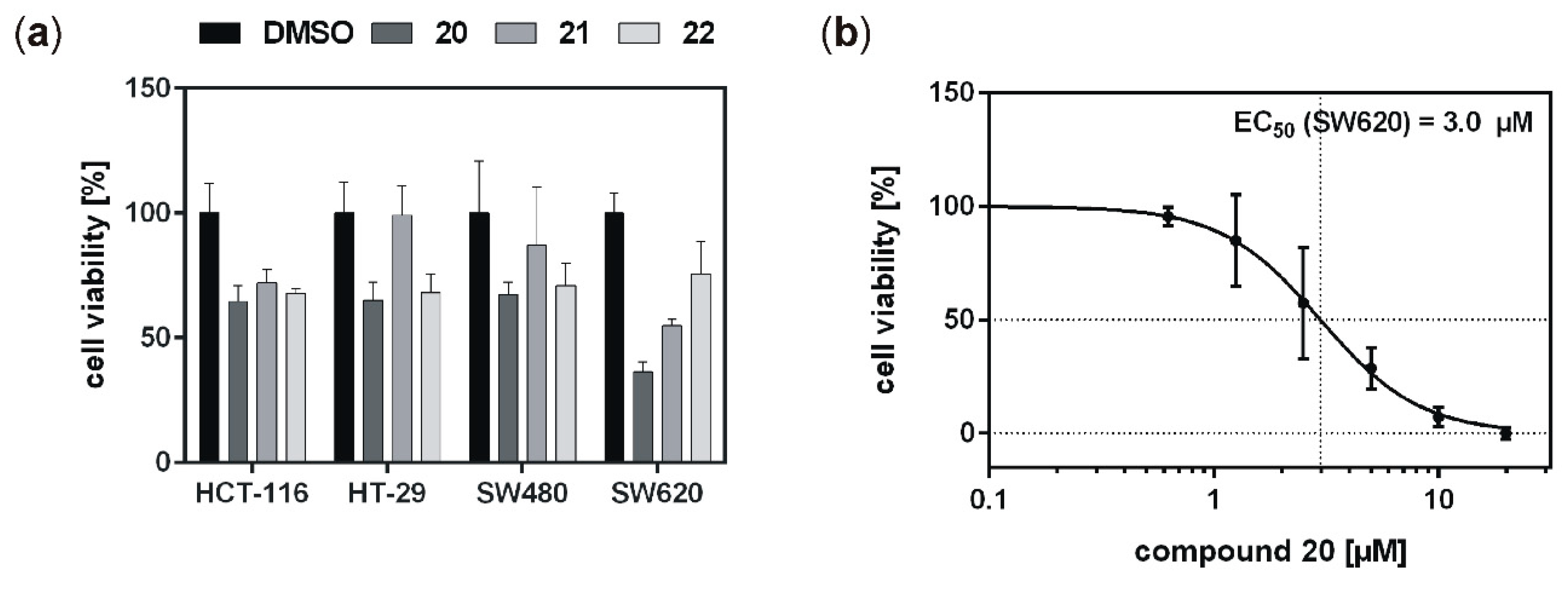
With the aim to identify more potent CK1δ-specific inhibitors, further IWP-derived compounds of a second set were also analyzed. Compared to the first tested set, all of these compounds resulted in remarkable inhibition of CK1δ (and ε), and determined IC
50 values for CK1δ were at least ten orders of magnitude lower than for compounds
16,
17,
18, or
19. In comparison to the originally tested IWP-compounds (IWP-2 and IWP-4; [
15]), inhibitors
20,
21, and
22 could be significantly improved concerning their inhibitory effects on CK1δ. Because highly potent inhibitors of CK1 isoforms could be used for the treatment of proliferative diseases, the highly potent inhibitors
20,
21, and
22 were also tested for their effects on established tumor cell lines. Previously, CK1-specific inhibitors already showed promising effects on colon cancer cell lines and therefore, also inhibitors
20,
21, and
22 were subsequently tested on colon cancer cell lines HCT-116, HT-29, SW480, and SW620. While the mutation status of CK1δ (and the highly related isoform ε) in these cell lines remains to be determined and confirmed, in a previous study, we already demonstrated robust expression of CK1δ and ε as well as CK1-specific kinase activity in SW480 and SW620 cells, while HT-29 cells showed remarkably reduced expression of CK1δ as well as reduced CK1-specific kinase activity [
2]. Consequently, cell line-specific differences could be observed for the tested inhibitors in our current study. The effects obtained for the treatment of HCT-116, HT-29, and SW480 cells were quite similar, although low expression of CK1δ has previously been demonstrated for HT-29 [
2]. The reduction of cell viability could therefore also be mediated via the inhibition of other CK1 isoforms or targeting of other proteins in general. Furthermore, in comparison to previously described difluoro-dioxolo-benzoimidazole-derived compounds like Richter-2 [
17], but also in comparison to the original IWP-compound IWP-4 [
15], the cellular effects mediated by inhibitors
20,
21, and
22 fell short of expectations. Inadequate ability to cross the cell membrane or low target affinity within the cell might be possible reasons for the obtained results. In contrast to
21 and
22, at least treatment with compound
20 led to comparably strong effects on viability of SW620 colon cancer cells. Keeping in mind that cell lines SW480 and SW620 derived from primary tumor (SW480) and metastasis (SW620) of the same patient, this finding is of particular interest [
37]. SW480 and SW620 represent different stages of colon cancer development, and in our investigation compound
20 showed stronger effects on the metastasis compared to the primary tumor. Survival of colon and rectal cancer patients is increasing continuously and, since the prognosis of patients is mainly determined by the occurrence of liver and lung metastases with a reported 5-year overall survival of only 12% [
18], successful treatment of distant metastases is desperately warranted to further improve outcome of colon and rectal cancer. With this regard, compound
20 may be an attractive agent for systemic treatment of metastatic disease. Individual screening and molecular subtyping may help in the future to identify possible candidates for such a targeted therapy [
19,
20].
In order to develop novel treatment strategies, detailed characterization of CK1δ (mutant)-associated functions will enable for (i) the deepened understanding of CK1δ-driven pathogenic processes and (ii) the design of new mutant-specific inhibitors with decreased off-target effects for being used in potential future therapies for various tumor entities, including colon and rectal cancer.
4. Materials and Methods
4.1. Generation of Expression Vectors
In order to introduce point mutations in CK1δ, site-directed mutagenesis using QuikChange II Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA, USA) was performed on pGEX6-P3 or pcDNA3.1 expression vectors, respectively, coding for GST-CK1δ or His-CK1δ. Mutagenesis was performed according to the manufacturer’s instructions using the primers listed in
Table S2. Generation of pcDNA3.1 expression vectors coding for His-CK1δ wild type or T67S mutant was described by Richter and colleagues [
2].
4.2. Expression and Purification of GST Fusion Proteins
Overexpression and purification of GST-CK1δ fusion proteins, either wild type or mutant, were performed as described previously [
38].
4.3. Kinase Assays
Each kinase assay reaction was performed in kinase buffer (25 mM Tris-HCl (pH 7.5), 10 μM ATP, 10 mM MgCl2, 100 μM EDTA) in presence of purified GST-CK1δ kinase as source of enzyme (either wild type or mutant), an assay-specific substrate (α-casein, GST-p531−64, or GST-β-catenin1−181), and radiolabeled ATP (2 μCi [γ-32P]-ATP). For performing Michaelis–Menten analysis increasing substrate concentrations were used (α-casein: 0.39−25 μM; GST-p531−64: 0.09−1.87 μM; GST-β-catenin1−181: 0.07−1.32 μM). For biological evaluation of small molecule inhibitors (SMIs), each inhibitor compound was either tested at 10 μM concentration for initial screening or by using a dilution series (0.005−10 µM) for determination of IC50 values. DMSO was used as control. Reactions were incubated for 30 min at 30 °C and reactions were stopped using 5x SDS-loading buffer (250 mM Tris-HCl [pH 6.8], 25% (v/v) β-mercaptoethanol (MSH), 50% (v/v) glycerol, 10% (w/v) SDS, 0.5% (w/v) bromphenol blue). Subsequently, samples were separated by SDS-PAGE using 12.5% acrylamide gels, stained with Coomassie, and phosphorylated substrates were visualized on X-ray film. Incorporation of radiolabeled phosphate was quantified by scintillation counting of Coomassie-stained substrate bands. Defined dilutions of the reaction master mix were counted as well in order to convert results obtained as counts per minute (CPM) to units of transferred phosphate in pmol/min. Michaelis–Menten analysis was performed using GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA).
4.4. Synthesis
All chemical reagents were commercially available from abcr GmbH, Karlsruhe, Germany; Sigma-Aldrich Chemie GmbH, Merck Group, Munich, Germany; Merck Millipore, Darmstadt, Germany; Acros Organics, Thermo Fisher Scientific, Geel, Belgium; TCI Deutschland GmbH, Eschborn, Germany; Honeywell, Morristown, NJ, USA; Grüssing GmbH, Filsum, Germany. Purchased chemicals were used without further purification. Infrared spectra (IR) were recorded on a Shimadzu IRAffinity-1S FTIR-spectrometer (Shimadzu Deutschland GmbH, Duisburg, Germany). NMR spectra were recorded on a Bruker Avance III 300 spectrometer (Bruker Corporation, Billerica, MA, USA) (300 MHz 1H frequency), (75 MHz 13C NMR frequency) and a Bruker Avance III 400 spectrometer (Bruker Corporation, Billerica, MA, USA) (400 MHz 1H frequency), (100 MHz 13C NMR frequency). Chemical shifts were reported in ppm, multiplicity, and coupling constant J (Hz). Spectra were referenced to internal DMSO-d6. Whenever appropriate, signal assignments were deduced from COSY, and CH correlation NMR experiments. LC-MS were recorded on a Bruker Esquire LC ion trap mass spectrometer spectrometer (ESI Bruker Corporation, Billerica, MA, USA). High resolution MS was performed on a Thermo Fischer Q Exactive PlusMS Hybrid Quadrupol-Orbitrap ESI-mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Column chromatography was performed using a LaFlash system (VWR) with Macherey–Nagel silica gel 60 (63−200 μm) for pre-columns and pre-packed Interchim PuriFlash-30SIHP silica gel columns (30 μM, 40 g) using mixtures of petroleum ether (PE) and ethyl acetate (EA). HPLC analysis was performed on a Hewlett-Packard 1050 Series using a STAGROMA YMC-C-18 column. All test compounds were proven by HPLC to have ≥97% purity. Where necessary, reactions were carried out in a nitrogen atmosphere using dry solvents.
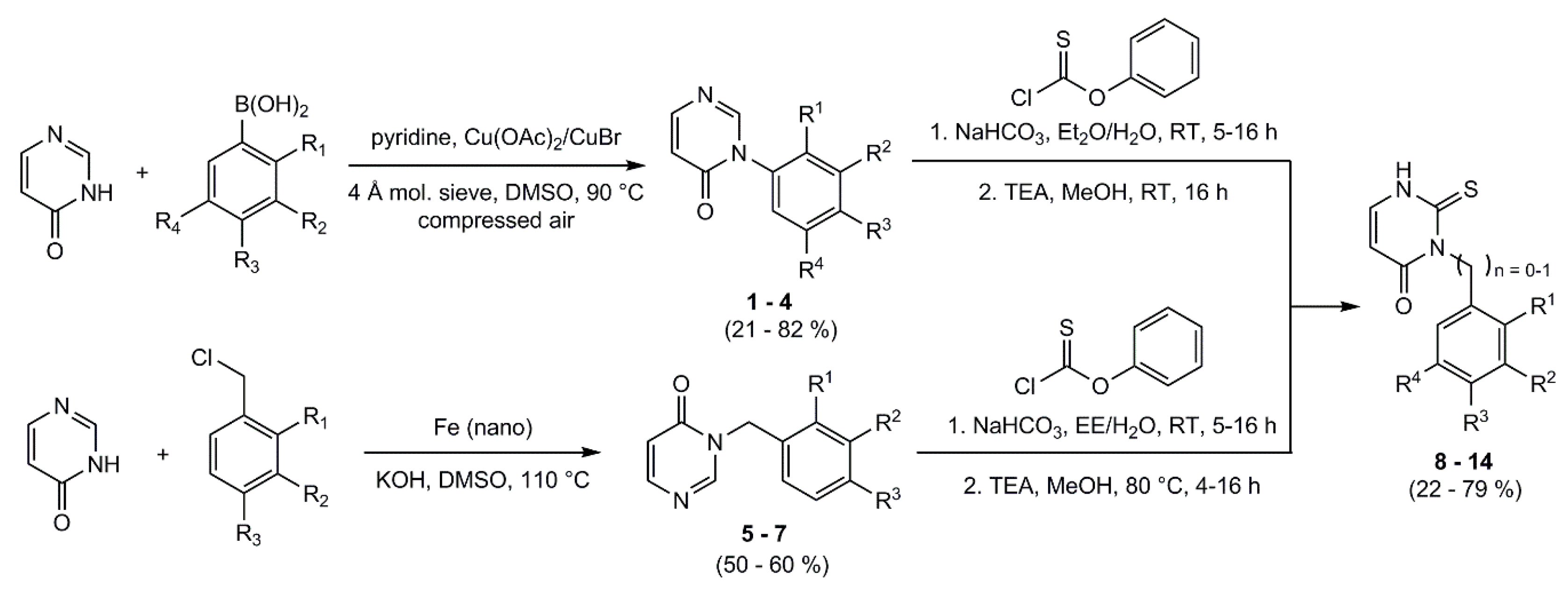
4.5. General Procedure for the Synthesis of the Phenyl Pyrimidinone-Derivatives
Molecular sieve (4 Å) was dried in vacuo. 4-(3H)-pyrimidinone, the appropriate phenylboronic acid, and a copper catalyst were dissolved in DMSO and pyridine was added. The reaction mixture was heated to 90 °C for 4 h and compressed air was added through a small tube into the reaction mixture. After cooling, the molecular sieve and the catalyst were filtered off; the filtrate was then quenched with water and extracted with ethyl acetate. The organic phase was washed with water and brine, dried over sodium sulfate, and the solvent was removed under reduced pressure. If necessary, the product was purified by column flash-chromatography on silica gel.
3-Phenylpyrimidin-4(3
H)-one (
1): 3-Phenylpyrimidin-4(3
H)-one (
1) was obtained from 4(3
H)-pyrimidinone (193 mg, 2.01 mmol), phenylboronic acid (487 mg, 3.99 mmol), copper(II)-acetate (72.0 mg, 0.40 mmol), and pyridine (330 μL, 4.05 mmol) in DMSO (32 mL) as brown solid. Yield: 260 mg (1.51 mmol; 76%). C
10H
8N
2O (M
r 172.19).
1H NMR (DMSO-
d6): δ = 8.44 (s, 1 H, C
2HPyr), 7.99 (dd,
3J = 6.7 Hz,
5J = 0.5 Hz, 1 H, C
6HPyr), 7.59–7.45 (m, 5 H, C
HPhen), 6.52 (dd,
3J = 6.7 Hz,
4J = 1.0 Hz, 1 H, C
5HPyr) ppm.
13C NMR (DMSO-
d6): δ = 159.8 (
C4Pyr), 153.5 (
C6Pyr), 152.0 (
C2Pyr), 137.1 (
C1Phen), 129.2 (
C3/5Phen), 128.9 (
C4Phen), 127.1 (
C2/6Phen), 115.6 (
C5Pyr) ppm. MS (ESI, 70 eV)
m/
z = 172.9 [M + H]
+. Synthesis has been described by Abd El Kader et al. [
26].
3-(2,5-Dimethoxyphenyl)pyrimidin-4(3H)-one (2): 3-(2,5-Dimethoxyphenyl)pyrimidin-4(3H)-one (2) was obtained from 4(3H)-pyrimidin-one (200 mg, 2.08 mmol), 2,5-dimethoxyphenylboronic acid (735 mg, 4.04 mmol), copper(I)-bromide (77.0 mg, 0.54 mmol), and pyridine (330 μL, 4.05 mmol) in DMSO (32 mL). Purification by column flash-chromatography on silica gel (gradient EE/PE) provided 2 as colorless solid. Yield: 202 mg (0.87 mmol, 42%). C12H12N2O3 (Mr 232.24). 1H NMR (DMSO-d6): δ = 8.30 (dd, 4J = 0.9 Hz, 5J = 0.6 Hz, 1 H, C2HPyr), 7.96 (dd, 3J = 6.7 Hz, 5J = 0.6 Hz, 1 H, C6HPyr), 7.19–7.15 (m, 1 H, C6HPhen), 7.09–7.05 (m, 2 H, C3HPhen, C4HPhen), 6.49 (dd, 3J = 6.7 Hz, 5J = 1.0 Hz, 1 H, C5HPyr), 3.74 (s, 3 H, C2-OCH3), 3.71 (s, 3 H, C5-OCH3) ppm. 13C NMR (DMSO-d6): δ = 159.5 (C4Pyr), 153.5 (C6Pyr), 153.0 (C2Phen), 152.8 (C2Pyr), 148.2 (C5Phen), 126.0 (C1Phen), 115.70 (C5Pyr), 115.66 (C3Phen), 114.8 (C4Phen), 113.5 (C6Phen), 56.3 (C2Phen-OCH3), 55.7 (C5Phen-OCH3) ppm. MS (ESI, 70 eV) m/z = 172.9 [M+H]+.
3-(2,4-Dimethoxyphenyl)pyrimidin-4(3H)-one (3): 3-(2,4-Dimethoxyphenyl)pyrimidin-4(3H)-one (3) was obtained from 4(3H)-pyrimidin-one (206 mg, 2.14 mmol), 2,4-dimethoxyphenylboronic acid (732 mg, 4.02 mmol), copper(I)-bromide (65.0 mg, 0.45 mmol) and pyridine (330 μL, 4.05 mmol) in DMSO (32 mL). Purification by column flash-chromatography on silica gel (gradient EE/PE) provided 3 as light brown solid. Yield: 102 mg (0.44 mmol, 21%). C12H12N2O3 (Mr 232.24). 1H NMR (DMSO-d6): δ = 8.25 (bs, 1 H, C2HPyr), 7.95 (dd, 3J = 6.7 Hz, 5J = 0.4 Hz, 1 H, C6HPyr), 7.27 (d, 3J = 8.6 Hz, 1 H, C6HPhen), 6.76 (d, 4J = 2.9 Hz, 1 H, C3HPhen), 6.64 (dd, 3J = 8.6 Hz, 4J = 2.6 Hz, 1 H, C5HPhen), 6.46 (dd, 3J = 6.7 Hz, 5J = 1.0 Hz, 1 H, C5HPyr), 3.83 (s, 3 H, C4-OCH3), 3.76 (s, 3 H, C2-CH3) ppm. 13C NMR (DMSO-d6): δ = 161.7 (C4Phen), 160.3 (C4Pyr), 155.6 (C2Phen), 153.9 (C6Pyr), 153.8 (C2Pyr), 130.0 (C6Phen), 119.1 (C1Phen), 116.1 (C5Pyr), 105.6 (C5Phen), 99.8 (C3Phen), 55.5 (C2-OCH3), 55.1 (C4-OCH3) ppm. MS (ESI, HR): [M + H]+ m/z = calc.: 233.09207 , found: 233.09200.
3-(3,5-Dimethoxyphenyl)pyrimidin-4(3H)-one (4): 3-(3,5-Dimethoxyphenyl)pyrimidin-4(3H)-one (4) was obtained from 4(3H)-pyrimidin-one (193 mg, 2.01 mmol), 3,5-dimethoxyphenylboronic acid (727 mg, 3.99 mmol), copper(I)-bromide (60.0 mg, 0.42 mmol) and pyridine (330 μL, 4.05 mmol) in DMSO (32 mL). Purification by column flash-chromatography on silica gel (gradient EE/PE) provided 4 as colorless solid. Yield: 381 mg (1.64 mmol, 82%). C12H12N2O3 (Mr 232.24). 1H NMR (DMSO-d6): δ = 8.41 (bs, 1 H, C2HPyr), 7.98 (dd, 3J = 6.7 Hz, 4J = 0.5 Hz, 1 H, C6HPyr), 6.67 (d, 4J = 2.2 Hz, 2 H, C2/6HPhen), 6.64 (t, 4J = 2.2 Hz, 1 H, C4HPhen), 6.50 (dd, 3J = 6.7 Hz, 5J = 1.0 Hz, 1 H, C5HPyr), 3.78 (s, 6 H, OCH3) ppm. 13C NMR (DMSO-d6): δ = 161.1 (C3/5Phen), 160.1 (C4Pyr), 153.9 (C6Pyr), 152.4 (C2Pyr), 139.2 (C1Phen), 116.2 (C5Pyr), 106.1 (C2/6Phen), 101.3 (C4Phen), 56.1 (OCH3) ppm. MS (ESI, HR): [M + H]+ m/z = calc.: 232.09207, found: 233.09200.
4.6. General Procedure for the Synthesis of the Benzyl Pyrimidinone-Derivatives
4(3H)-Pyrimidinone and potassium hydroxide were dissolved in DMSO. Under nitrogen-atmosphere iron (nano) and the appropriate benzyl chloride were added. The mixture was heated at 110 °C for 2 h, decanted in ice and acidified with 1 M HCl. The aqueous phase was extracted with DCM. The organic phase was washed with water, dried over sodium sulfate and the solvent was removed under reduced pressure. The crude products were purified by column flash-chromatography on silica gel (gradient EE/PE).
3-(4-Methoxybenzyl)pyrimidin-4(3H)-one (5): 3-(4-Methoxybenzyl)pyrimidin-4(3H)-one (5) was obtained from 4(3H)-pyrimidinone (240 mg, 2.50 mmol), potassium hydroxide (213 mg, 3.79 mmol), nano-iron (7.2 mg, 0.13 mmol) and 1-(chloromethyl)-4-methoxybenzene (350 µL, 2.50 mmol) in DMSO (3 mL). Purification by column flash-chromatography on silica gel (gradient EE/PE) provided 5 as yellow solid. Yield: 270 mg (1.25 mmol, 50%). C12H12N2O2 (Mr 216.24). 1H NMR (DMSO-d6): δ = 8.66 (s, 1 H, C2HPyr), 7.90 (d, 3J = 6.6 Hz, 1 H, C6HPyr), 7.31 (d, 3J = 6.6 Hz, 2 H, C2/6HBenz), 6.90 (d, 3J = 6.9 Hz, 2 H, C3/5HBenz), 6.40 (d, 3J = 6.4 Hz, 1 H, C5HPyr), 5.01 (s, 2 H, CH2), 3.72 (s, 3 H, OCH3) ppm. 13C NMR (DMSO-d6): δ = 160.0 (C4Pyr), 159.9 (C4Benz), 153.6 (C6Pyr), 152.7 (C2Pyr), 129.6 (C2/6Benz), 128.4 (C1Benz), 115.2 (C5Pyr), 114.0 (C3/5Benz), 55.1 (OCH3), 48.4 (CH2) ppm. MS (ESI, 70 eV) m/z = 217.1 [M + H]+.
3-(4-Trifluoromethylbenzyl)pyrimidin-4(3H)-one (6): 3-(4-Trifluoromethylbenzyl)pyrimidin-4(3H)-one (6) was obtained from 4(3H)-pyrimidinone (247 mg, 2.57 mmol), potassium hydroxide (223 mg, 3.97 mmol), nano-iron (7.4 mg, 0.13 mmol) and 1-(chloromethyl)-4-(trifluoromethyl)benzene (370 µL, 2.50 mmol) in DMSO (2.5 mL). Purification by column flash-chromatography on silica gel (gradient EE/PE) provided 6 as yellow solid. Yield: 383 mg (1.51 mmol, 60%). C12H9F3N2O (Mr 254.21). 1H NMR (DMSO-d6): δ = 8.71 (s, 1 H, C2HPyr), 7.96 (dd, 3J = 6.6 Hz, 4J = 0.4 Hz, 1 H, C6HPyr), 7.73 (d, 3J = 8.1 Hz, 2 H, C3/5HBenz), 7.52 (d, 3J = 8.1 Hz, 2 H, C2/6HBenz), 6.45 (dd, 3J = 6.6 Hz, 5J = 0.9 Hz, 1 H, C5HPyr), 5.19 (s, 2 H, CH2), ppm. 13C NMR (DMSO-d6): δ = 160.0 (C4Pyr), 153.9 (C6Pyr), 153.0 (C2Pyr), 141.1 (C1Benz), 128.3 (C2/6Benz), 128.3 (d, 2JCF = 31.4 Hz, C4Benz), 125.6 (q, 3JCF = 3.9 Hz, C3/5Benz), 124.2 (d, 1JCF = 271.5 Hz, CF3), 115.3 (C5Pyr), 48.8 (CH2) ppm. MS (ESI, 70 eV) m/z = 255.0 [M + H]+.
3-(2-Fluorobenzyl)pyrimidin-4(3H)-one (7): 3-(2-Fluorobenzyl)pyrimidin-4(3H)-one (7) was obtained from 4(3H)-pyrimidinone (247 mg, 2.57 mmol), potassium hydroxide (228 mg, 4.06 mmol), nano-iron (8.1 mg, 0.15 mmol) and 1-(chloromethyl)-2-fluorobenzene (300 µL, 2.50 mmol) in DMSO (3 mL). Purification by column flash-chromatography on silica gel (gradient EE/PE) provided 7 as yellow solid. Yield: 266 mg (1.31 mmol, 51%). C11H9FN2O (Mr 204.20). 1H NMR (DMSO-d6): δ = 8.61 (s, 1 H, C2HPyr), 7.94 (d, 3J = 6.6 Hz, 1 H, C6HPyr), 7.39–7.33 (m, 1 H, C3HBenz), 7.24–7.15 (m, 3 H, C4HBenz, C5HBenz, C6HBenz), 6.42 (dd, 3J = 6.6 Hz, 5J = 0.8 Hz, 1 H, C5HPyr), 5.14 (s, 2 H, CH2), ppm. 13C NMR (DMSO-d6): δ = 160.2 (d, 1JCF = 245.5 Hz, C2Benz), 159.9 (C4Pyr), 153.8 (C6Pyr), 153.0 (d, 5JCF = 2.2 Hz, C2Pyr), 130.0 (d, 3JCF = 11.9 Hz, C6Benz), 130.0 (C5Benz), 124.6 (d, 3JCF = 3.5 Hz, C4Benz), 123.0 (d, 2JCF = 14.1 Hz, C1Benz), 115.6 (d, 2JCF = 20.9 Hz, C3Benz), 115.2 (C5Pyr), 44.0 (d, 3JCF = 4.0 Hz, CH2) ppm. MS (ESI, 70 eV) m/z = 205.1 [M + H]+.
4.7. General Procedure for the Synthesis of the Phenyl/benzyl-2-thioxo-Pyrimidinone-Derivatives
The appropriate phenyl/benzyl- pyrimidinone derivative and NaHCO3 were dissolved in a mixture of organic solvent and deionized water under vigorous stirring. Phenyl thionochloroformate was added dropwise and the mixture was stirred at room temperature for 5–16 h. After extraction with ethyl acetate the organic phase was washed three times with brine, dried over sodium sulfate and the solvent was removed under reduced pressure. The residue was then dissolved in methanol. TEA was added and the mixture was stirred at room temperature (or at 80 °C) for 4–16 h. The solvent was removed and the crude products were purified by column flash-chromatography on silica gel (gradient EE/PE).
3-Phenyl-2-thioxo-2,3-dihydropyrimidin-4(1
H)-one (
8): 3-Phenyl-2-thioxo-2,3-dihydropyrimidin-4(1
H)-one (
8) was obtained from
1 (176 mg, 1.03 mmol), NaHCO
3 (512 mg, 6.09 mmol) and phenyl thionochloroformate (300 µL, 2.15 mmol) in diethyl ether (3 mL) and water (3 mL) in the first step. The residue was dissolved in methanol (6 mL) at room temperature (RT) and TEA (430 µL, 3.10 mmol) was added. Purification by column flash-chromatography on silica gel (gradient EE/PE) provided
8 as yellow solid. Yield: 95.8 mg (0.47 mmol, 46%). C
10H
8N
2OS (M
r 204.25). MS (ESI, 70 eV)
m/
z = 172.9 [M + H]
+. Synthesis has been described by Nekkaa et al. [
29].
3-(2,5-Dimethoxyphenyl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (9): 3-(2,5-Dimethoxyphenyl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (9) was obtained from 2 (244 mg, 1.05 mmol), NaHCO3 (536 mg, 6.38 mmol) and phenyl thionochloroformate (370 µL, 2.66 mmol) in diethyl ether (3 mL), ethyl acetate (3 mL) and water (3 mL) in the first step. The residue was dissolved in methanol (6 mL) at RT and TEA (451 µL, 3.25 mmol) was added. Purification by column flash-chromatography on silica gel (gradient EE/PE) provided 9 as light yellow solid. Yield: 209 mg (0.79 mmol, 79%). C12H12N2O3S (Mr 264.30). 1H NMR (DMSO-d6): δ = 12.59 (d, 3J = 4.9 Hz, 1 H, N-H), 7.51 (dd, 3J = 7.6 Hz, 3J = 5.8 Hz, 1 H, C6HPyr), 7.06 (d, 3J = 9.0 Hz, 1 H, C3HPhen), 6.95 (dd, 3J = 9.0 Hz, 4J = 3.0 Hz, 1 H, C4HPhen), 6.77 (d, 4J = 3.0 Hz, 1 H, C6HPhen), 5.99 (d, 3J = 7.6 Hz, 1 H, C5HPyr), 3.71 (s, 3 H, C5-OCH3), 3.67 (s, 3 H, C2-OCH3) ppm. 13C NMR (DMSO-d6): δ = 178.0 (C2Pyr), 160.6 (C4Pyr), 153.7 (C5Phen), 148.8 (C2Phen), 141.8 (C6HPyr), 128.5 (C1Phen), 116.1 (C6Phen), 114.8 (C4Phen), 113.7 (C3Phen), 105.0 (C5Pyr), 56.6 (C2-OCH3), 56.0 (C5-OCH3) ppm. MS (ESI, 70 eV) m/z = 265.0 [M + H]+, 529.0 [MMH]+.
3-(2,4-Dimethoxyphenyl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (10): 3-(2,4-Dimethoxyphenyl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (10) was obtained from 3 (125 mg, 0.54 mmol), NaHCO3 (278 mg, 3.31 mmol) and phenyl thionochloroformate (194 µL, 1.39 mmol) in diethyl ether (3 mL), ethyl acetate (3 mL) and water (3 mL) in the first step. The residue was dissolved in methanol (6 mL) at RT and TEA (237 µL, 1.71 mmol) was added. Purification by column flash-chromatography on silica gel (gradient EE/PE) provided 10 as light yellow solid. Yield: 114 mg (0.43 mmol, 78%). C12H12N2O3S (Mr 264.30). 1H NMR (DMSO-d6): δ = 12.54 (s, 1 H, N-H), 7.48 (d, 3J = 7.6 Hz, 1 H, C6HPyr), 7.00 (d, 3J = 8.6 Hz, 1 H, C6HPhen), 6.66 (d, 4J = 2.6 Hz, 1 H, C3HPhen), 6.56 (dd, 3J = 8.6 Hz, 4J = 2.6 Hz, 1 H, C5HPhen), 5.97 (d, 3J = 7.6 Hz, 1 H, C5HPyr), 3.80 (s, 3 H, C2-OCH3), 3.70 (s, 3 H, C4-OCH3) ppm. 13C NMR (DMSO-d6): δ = 178.1 (C2Pyr), 160.4 (C4Pyr), 160.4 (C2Phen), 154.9 (C4Phen), 141.2 (C6Pyr), 129.9 (C6Phen), 120.5 (C1Phen), 105.1 (C5Phen), 104.4 (C5Pyr), 99.3 (C3Phen), 55.7 (C4-OCH3), 55.4 (C2-OCH3) ppm. MS (ESI, 70 eV) m/z = 265.0 [M + H]+, 529.0 [MMH]+.
3-(3,5-Dimethoxyphenyl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (11): 3-(3,5-Dimethoxyphenyl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (11) was obtained from 4 (350 mg, 1.51 mmol), NaHCO3 (759 mg, 9.03 mmol) and phenyl thionochloroformate (529 µL, 3.80 mmol) in diethyl ether (6 mL) and water (6 mL) in the first step. The residue was dissolved in methanol (15 mL) at RT and TEA (645 µL, 4.65 mmol) was added. Purification by column flash-chromatography on silica gel (gradient EE/PE) provided 11 as yellow solid. Yield: 280 mg (1.06 mmol, 70%). C12H12N2O3S (Mr 264.30). 1H NMR (DMSO-d6): δ = 12.62 (d, 3J = 3.1 Hz, 1 H, N-H), 7.50 (d, 3J = 7.6 Hz, 3J = 5.1 Hz, 1 H, C6HPyr), 6.52 (t, 4J = 2.3 Hz, 1 H, C4HPhen), 6.38 (d, 4J = 2.3 Hz, 2 H, C2/6HPhen), 6.01 (d, 3J = 7.6 Hz, 1 H, C5HPyr), 3.73 (s, 6 H, C3/5-OCH3) ppm. 13C NMR (DMSO-d6): δ = 177.5 (C2Pyr), 160.8 (C3/5Phen), 160.5 (C4Pyr), 141.2 (C6Pyr), 140.7 (C1Phen), 106.9 (C2/6Phen), 104.9 (C5Pyr), 100.0 (C4Phen), 55.4 (C3/5-OCH3) ppm. MS (ESI, 70 eV) m/z = 265.0 [M + H]+, 529.0 [MMH]+.
3-(4-Methoxybenzyl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (12): 3-(4-Methoxybenzyl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (12) was obtained from 5 (251 mg, 1.16 mmol), NaHCO3 (586 mg, 6.97 mmol) and phenyl thionochloroformate (380 µL, 2.73 mmol) in EE (5 mL) and water (5 mL) in the first step. The residue was dissolved in methanol (10 mL) and TEA (429 µL, 3.09 mmol) was added. The mixture was stirred at 80 °C for 9 h. Purification by column flash-chromatography on silica gel (gradient EE/PE) provided 12 as colorless solid. Yield: 65.5 mg (0.26 mmol, 23%). C12H12N2O2S (Mr 248.30). 1H NMR (DMSO-d6): δ = 12.63 (bs, 1 H, N-H), 7.47 (dd, 3J = 7.5 Hz, 3J = 5.7 Hz, 1 H, C6HPyr), 7.28 (d, 3J = 8.7 Hz, 2 H, C2/6HBenz), 6.85 (d, 3J = 8.8 Hz, 2 H, C3/5HBenz), 5.97 (dd, 3J = 7.5 Hz, 4J = 0.7 Hz, 1 H, C5HPyr), 5.43 (s, 2 H, CH2), 3.71 (s, 3 H, OCH3) ppm. 13C NMR (DMSO-d6): δ = 176.9 (C2Pyr), 160.4 (C4Pyr), 158.1 (C4Benz), 140.9 (C6Pyr), 129.1 (C2/6Benz), 128.1 (C1Benz), 113.6 (C3/5Benz), 104.4 (C5Pyr), 55.1 (OCH3), 47.7 (CH2) ppm. MS (ESI, 70 eV) m/z = 248.9 [M + H]+.
3-(4-Trifluoromethylbenzyl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (13): 3-(4-Trifluoromethylbenzyl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (13) was obtained from 6 (298 mg, 1.17 mmol), NaHCO3 (597 mg, 7.11 mmol) and phenyl thionochloroformate (380 µL, 2.73 mmol) in EE (5 mL) and water (5 mL) in the first step. The residue was dissolved in methanol (10 mL) and TEA (433 µL, 3.12 mmol) was added. The mixture was stirred at 80 °C for 4 h. Purification by column flash-chromatography on silica gel (gradient EE/PE) provided 13 as yellow solid. Yield: 142 mg (0.50 mmol, 42%). C12H9F3N2OS (Mr 286.27). 1H NMR (DMSO-d6): δ = 12.75 (d, 3J = 5.2 Hz, 1 H, N-H), 7.67 (d, 3J = 8.1 Hz, 2 H, C3/5HBenz), 7.54 (dd, 3J = 7.5 Hz, 3J = 5.8 Hz, 1 H, C6HPyr), 7.47 (d, 3J = 8.0 Hz, 2 H, C2/6HBenz), 6.02 (dd, 3J = 7.5 Hz, 4J = 1.4 Hz, 1 H, C5HPyr), 5.58 (s, 2 H, CH2) ppm. 13C NMR (DMSO-d6): δ = 177.1 (C2Pyr), 160.4 (C4Pyr), 141.3 (C6Pyr), 141.1 (C1Benz), 127.9 (d, 2JCF = 35.2 Hz, C4Benz), 128.1 (C1Benz), 127.7 (C2/6Benz), 125.2 (q, 3JCF = 3.8 Hz, C3/5Benz), 124.3 (d, 1JCF = 272.3 Hz, CF3), 104.3 (C5Pyr), 48.2 (CH2) ppm. MS (ESI, 70 eV) m/z = 286.9 [M + H]+.
3-(2-Fluorobenzyl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (14): 3-(2-Fluorobenzyl)-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (14) was obtained from 7 (223 mg, 1.09 mmol), NaHCO3 (552 mg, 6.57 mmol) and phenyl thionochloroformate (360 µL, 2.59 mmol) in EE (5 mL) and water (5 mL) in the first step. The residue was dissolved in methanol (8 mL) and TEA (402 µL, 2.90 mmol) was added. The mixture was stirred at 80 °C for 16 h. Purification by column flash-chromatography on silica gel (gradient EE/PE) provided 14 as colorless solid. Yield: 55.4 mg (0.23 mmol, 22%). C11H9FN2OS (Mr 236.26). 1H NMR (DMSO-d6): δ = 12.74 (d, 3J = 5.4 Hz, 1 H, N-H), 7.54 (dd, 3J = 7.5 Hz, 3J = 5.8 Hz, 1 H, C6HPyr), 7.32–7.26 (m, 1 H, C4HBenz), 7.22–7.12 (m, 1 H, C3HBenz), 7.11 (td, 3J = 7.5 Hz, 4J = 1.0 Hz, 1 H, C5HBenz), 7.00–6.97 (m, 1 H, C6HBenz), 6.02 (dd, 3J = 7.5 Hz, 4J = 1.3 Hz, 1 H, C5HPyr), 5.53 (s, 2 H, CH2) ppm. 13C NMR (DMSO-d6): δ = 177.1 (C2Pyr), 160.3 (C4Pyr), 159.9 (d, 1JCF = 244.3 Hz, C2Benz), 141.3 (C6Pyr), 128.8 (d, 3JCF = 8.1 Hz, C4Benz), 127.0 (d, 3JCF = 4.1 Hz, C6Benz), 124.4 (d, 4JCF = 3.4 Hz, C5Benz), 122.9 (d, 2JCF = 14.1 Hz, C1Benz), 115.2 (d, 2JCF = 21.0 Hz, C3Benz), 104.2 (C5Pyr), 42.8 (d, 3JCF = 5.5 Hz, CH2) ppm. MS (ESI, 70 eV) m/z = 237.0 [M + H]+.
4.8. Synthesis of the Benzothiazole–Linker
2-Chloro-
N-(6-(trifluoromethyl)benzo[d]thiazol-2-yl)acetamide (
15): A solution of 2-amino-6-(trifluoromethyl)benzo[
d]thiazole (542 mg, 2.48 mmol) and TEA (412 µL, 2.73 mmol) in DCM (4.5 mL) was added dropwise to a solution of 2-chloracetyl chloride (217 µL, 2.73 mmol) in DCM (1.5 mL). The mixture was then stirred for 24 h at rt. The solvent was concentrated under reduced pressure. The product was filtered, washed with water and recrystallized from EtOH/H
2O to give light brownish crystals. Yield: 585 mg (1.99 mmol, 80%). C
10H
6ClF
3N
2OS (M
r 294,68).
1H NMR (DMSO-
d6): δ = 12.96 (s, 1 H, N
H), 8.52 (d,
4J = 1.6 Hz, 1 H, C
7HBnth), 7.94 (d,
3J = 8.5 Hz, 1 H, C
4HBnth), 7.75 (dd,
3J = 8.5 Hz,
4J = 1.6 Hz, 1 H, C
5HBnth), 4.50 (s, 2 H, C
H2-Cl) ppm.
13C NMR (DMSO-
d6): δ = 166.4 (
C=O), 160.8 (C
2Bnth), 151.2 (
C3aBnth), 132.0 (
C7aBnth)
, 124.5 (d,
1JCF = 271.8 Hz,
CF
3), 123.9 (d,
2JCF = 31.9 Hz, C
6Bnth), 123.0 (d,
3JCF = 3.6 Hz,
C5H
Bnth), 121.2 (
C4H
Bnth), 120.0 (d,
3JCF = 4.0 Hz,
C7H
Bnth), 42.5 (
CH
2-Cl) ppm. MS (ESI, 70 eV)
m/
z = 294.8 [M + H]
+. Synthesis has been described by García-Reyes et al. [
15].
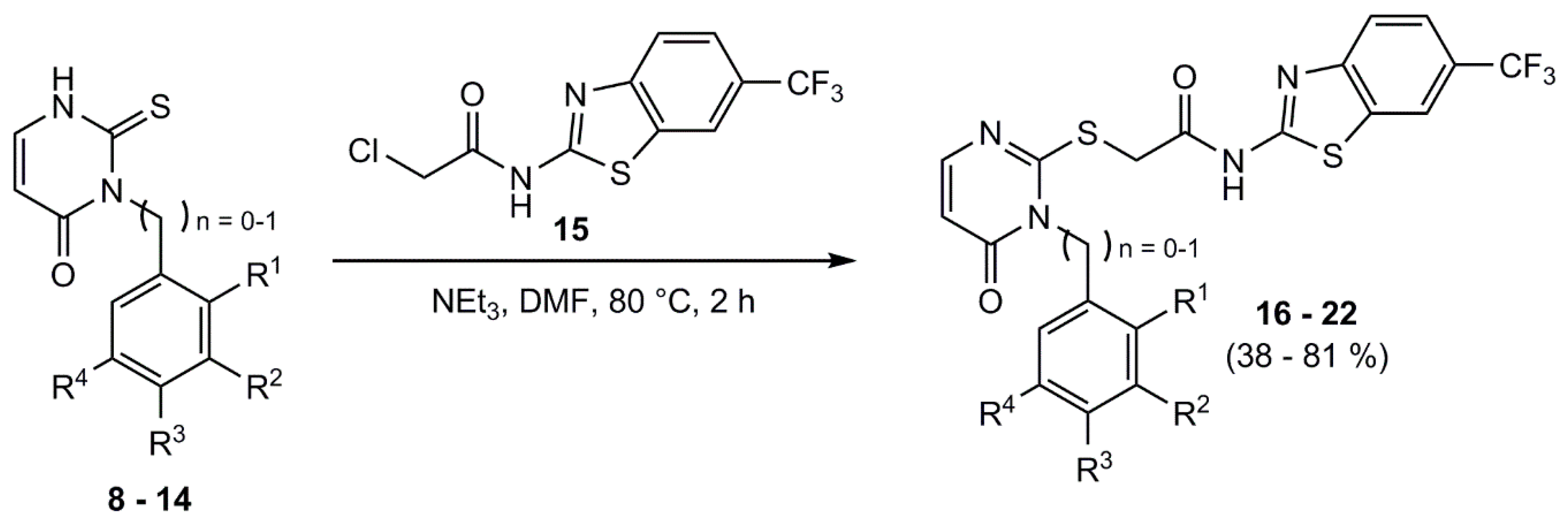
4.9. General Procedure for the Synthesis of the IWP Derived Derivatives
Under nitrogen atmosphere the appropriate phenyl/benzyl-2-thioxo-pyrimidinone-derivative and 15 were dissolved in DMF. TEA was added and the mixture was heated to 80 °C for 2 h. After quenching with water, the reaction mixture was extracted with ethyl acetate. The organic phase was washed respectively with deionized water and brine three times and dried over sodium sulfate. The solvent was removed under reduced pressure and the crude product was purified by column flash-chromatography on silica gel (gradient EE/PE) or by recrystallization from EtOH/H2O.
2-((3-Phenyl-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoromethyl)benzo-[d]thiazol-2-yl)acetamide (16): 2-((3-Phenyl-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoromethyl)benzo-[d]thiazol-2-yl)acetamide (16) was obtained from 15 (146 mg, 0.50), 8 (131 mg, 0.50 mmol), and TEA (0.2 mL, 1.44 mmol). Purification by chromatography and recrystallization from EtOH/H2O provided 16 as light yellow solid. Yield: 102 mg (0.22 mmol, 44%). C20H13F3N4O2S2 (Mr 462.47). HPLC (purity): 100%. 1H NMR (DMSO-d6): δ = 12.87 (s, 1 H, NH), 8.49 (s, 1 H, C7HBnth), 7.92 (d, 3J = 8.6 Hz, 1 H, C4HBnth), 7.85 (d, 3J = 6.6 Hz, 1 H, C6HPyr), 7.75 (d, 3J = 8.6 Hz, 1 H, C5HBnth), 7.63–7.58 (m, 3 H, C4HPhen, C3HPhen), 7.45–7.43 (m, 2 H, C2HPhen), 6.29 (d, 3J = 6.6 Hz, 1 H, C5HPyr), 4.21 (s, 2 H, CH2-C=O) ppm. 13C NMR (DMSO-d6): δ = 167.3 (CH2-C=O), 161.0 (C2Bnth), 160.7 (C4Pyr), 152.3 (C6Pyr), 151.3 (C3aBnth), 135.5 (C1Phen), 132.0 (C7aBnth), 130.1 (C4HPhen), 129.7 (C3HPhen), 128.7 (C2HPhen), 124.5 (d, 1JCF = 271.7 Hz, CF3), 123.8 (d, 2JCF = 31.9 Hz, C6Bnth), 123.0 (d, 3JCF = 3.1 Hz, C5HBnth), 121.0 (C4HBnth), 119.9 (d, 3JCF = 4.4 Hz, C7HBnth) 111.0 (C5Pyr), 36.0 (CH2-C=O) ppm. MS (ESI, 70 eV) m/z = 463.0 [M + H]+. IR: = 3132, 3036, 2989, 1549, 1478, 1314, 1152, 1117, 1078, 833, 826, 698, 677, cm−1.
2-((3-(2,5-Dimethoxyphenyl)-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoromethyl)benzo[d]thiazol-2-yl)acetamide (17):2-((3-(2,5-Dimethoxyphenyl)-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoro-methyl)benzo[d]thiazol-2-yl)acetamide (17) was obtained from 15 (202 mg, 0.69 mmol), 9 (173 mg, 0.65 mmol), and TEA (0.27 mL, 1.95 mmol). Recrystallization from EtOH/H2O provided 17 as light yellow solid. Yield: 243 mg (0.46 mmol, 71%). C22H17F3N4O4S2 (Mr 522.52). HPLC (purity): 98%. 1H NMR (DMSO-d6): δ = 12.87 (s, 1 H, NH), 8.50 (s, 1 H, C7HBnth), 7.92 (d, 3J = 8.4 Hz, 1 H, C4HBnth), 7.83 (d, 3J = 6.6 Hz, 1 H, C6HPyr), 7.75 (d, 3J = 8.4 Hz, 1 H, C5HBnth), 7.22 (d, 3J = 9.1 Hz, 1 H, C3HPhen), 7.13 (dd, 3J = 9.1 Hz, 4J = 2.9 Hz, 1 H, C4HPhen), 7.04 (d, 4J = 2.9 Hz, 1 H, C6HPhen), 6.25 (d, 3J = 6.6 Hz, 1 H, C5HPyr), 4.20 (s, 2 H, CH2-C=O), 3.753 (s, 3H, CH3-OC2Phen), 3.745 (s, 3H, CH3-OC5Phenyl) ppm. 13C NMR (DMSO-d6): δ = 167.4 (CH2-C=O), 163.1 (C2Pyr), 161.0 (C2Bnth), 160.2 (C4Pyr), 153.4 (C5Phen), 152.4 (C6Pyr), 151.3 (C3aBnth), 148.6 (C2Phen), 132.0 (C7aBnth), 124.6 (d, 1JCF = 271.9 Hz, CF3), 124.1 (C1Phen), 123.8 (d, 2JCF = 31.8 Hz, C6Bnth), 123.0 (d, 3JCF = 3.3 Hz, C5HBnth), 121.1 (C4HBnth), 120.0 (d, 3JCF = 4.1 Hz, C7HBnth), 116.8 (C4HPhen), 115.6 (C6HPhen), 113.9 (C3Hphen), 110.8 (C5Pyr), 56.4 (CH3-OC5Phen), 55.8 (CH3-OC2Phen), 36.0 (CH2-C=O) ppm. MS (ESI, HR): [M + H]+ m/z = calc.: 523.072159, found.: 532.07159. IR: = 3138, 3059, 2968, 2930, 1665, 1553, 1510, 1481, 1317, 1269, 1153, 1101, 1078, 1038, 839, 831, 677 cm−1.
2-((3-(2,4-Dimethoxyphenyl)-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoromethyl)benzo[d]thiazol-2-yl)acetamide (18): 2-((3-(2,4-Dimethoxyphenyl)-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoro-methyl)benzo[d]thiazol-2-yl)acetamide (18) was obtained from 15 (117 mg, 0.40 mmol), 10 (93.3 mg, 0.35 mmol), and TEA (158 µL, 1.14 mmol). Purification by chromatography provided 18 as light yellow solid. Yield: 109 mg (0.21 mmol, 59%). C22H17F3N4O4S2 (Mr 522.52). HPLC (purity): 97%. 1H NMR (DMSO-d6): δ = 12.87 (s, 1 H, NH), 8.50 (bs, 1 H, C7HBnth), 7.92 (d, 3J = 8.5 Hz, 1 H, C4HBnth), 7.82 (d, 3J = 6.6 Hz, 1 H, C6HPyr), 7.75 (dd, 3J = 8.5 Hz, 4J = 1.5 Hz, 1 H, C5HBnth), 7.26 (d, 3J = 8.6 Hz, 1 H, C6HPhen), 6.80 (d, 4J = 2.5 Hz, 1 H, C3HPhen), 6.68 (dd, 3J = 8.6 Hz, 4J = 2.5 Hz, 1 H, C5HPhen), 6.23 (d, 3J = 6.6 Hz, 1 H, C5HPyr), 4.18 (d, 4J = 1.7 Hz, 2 H, CH2-C=O), 3.85 (s, 3H, CH3-OC4Phen), 3.78 (s, 3H, CH3-OC2Phen) ppm. 13C NMR (DMSO-d6): δ = 167.5 (CH2-C=O), 163.9 (C2Pyr), 162.1 (C4Phen), 161.0 (C2Bnth), 160.5 (C4Pyr), 155.6 (C2Phen), 152.4 (C6HPyr), 151.3 (C3aBnth), 132.0 (C7aBnth), 130.6 (C6HPhen), 124.6 (d, 1JCF = 271.8 Hz, CF3), 123.8 (d, 2JCF = 31.9 Hz, C6Bnth), 123.0 (d, 3JCF = 3.6 Hz, C5HBnth), 121.1 (C4HBnth), 120.0 (d, 3JCF = 4.2 Hz, C7HBnth), 116.5 (C1Phen), 110.87(C5Pyr), 105.8 (C5HPhen), 99.6 (C3Phen), 56.1 (CH3-OC2Phen), 55.7 (CH3-OC4Phen), 35.9 (CH2-C=O) ppm. MS (ESI, 70 eV) [M+H]+ m/z = calc.: 523.072159, found.: 532.07156. IR: = 3067, 2976, 2941, 1688, 1541, 1485, 1317, 1269, 1159, 1115, 1082, 1028, 824, 718, 677cm−1.
2-((3-(3,5-Dimethoxyphenyl)-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoromethyl)benzo[d]thiazol-2-yl)acetamide (19): 2-((3-(3,5-Dimethoxyphenyl)-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoro-methyl)benzo[d]thiazol-2-yl)acetamide (19) was obtained from 15 (223 mg, 0.76 mmol), 11 (185 mg, 0.70 mmol), and TEA (291 µL, 2.10 mmol). Purification by chromatography provided 19 as light yellow solid. Yield: 248 mg (0.48 mmol, 68%). C22H17F3N4O4S2 (Mr 522.52). HPLC (purity): 98%. 1H NMR (DMSO-d6): δ = 12.88 (s, 1 H, NH), 8.50 (bs, 1 H, C7HBnth), 7.92 (d, 3J = 8.5 Hz, 1 H, C4HBnth), 7.83 (d, 3J = 6.6 Hz, 1 H, C6HPyr), 7.75 (dd, 3J = 8.5 Hz, 4J = 1.1 Hz, 1 H, C5HBnth), 6.70 (t, 4J = 2.1 Hz, 1 H, C4HPhen), 6.64 (d, 4J = 2.1 Hz, 2 H, C2/6HPhen), 6.26 (d, 3J = 6.6 Hz, 1 H, C5HPyr), 4.20 (s, 2 H, CH2-C=O), 3.79 (s, 6H, CH3-OC) ppm. 13C NMR (DMSO-d6): δ = 167.5 (CH2-C=O), 162.5 (C2Pyr), 161.1 (C3/5Phen), 161.0 (C2Bnth), 160.5 (C4Pyr), 152.2 (C6HPyr), 151.3 (C3aBnth), 137.2 (C1Phen), 132.0 (C7aBnth), 124.6 (d, 1JCF = 272.3 Hz, CF3), 123.8 (d, 2JCF = 31.7 Hz, C6Bnth), 123.0 (d, 3JCF = 3.5 Hz, C5HBnth), 121.1 (C4HBnth), 120.0 (d, 3JCF = 4.2 Hz, C7HBnth), 111.0 (C5Pyr), 106.9 (C2/6HPhen), 101.7 (C4HPhen), 55.7 (CH3-OC3/5Phen), 36.0 (CH2-C=O) ppm. MS (ESI, 70 eV) m/z = 522.9 [M+H]+. IR: = 3070, 2957, 1668, 1511, 1498, 1317, 1269, 1153, 1111, , 839, 826, 679 cm−1.
2-((3-(4-Methoxybenzyl)-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoromethyl)benzo[d]thiazol-2-yl)acetamide (20):2-((3-(4-Methoxybenzyl)-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoromethyl)benzo[d]thiazol-2-yl)acetamide (20) was obtained from 15 (72.2 mg, 0.25 mmol), 12 (57.8 mg, 0.23 mmol), and TEA (96.9 µL, 0.70 mmol); 20 was washed with DCM and collected as colorless solid. Yield: 46.9 mg (92.6 µmol, 40%). C22H17F3N4O2S2 (Mr 506.52). HPLC (purity): 100%. 1H NMR (DMSO-d6): δ = 12.95 (s, 1 H, NH), 8.49 (bs, 1 H, C7HBnth), 7.92 (d, 3J = 8.4 Hz, 1 H, C4HBnth), 7.78 (d, 3J = 6.5 Hz, 1 H, C6HPyr), 7.74 (d, 3J = 8.6 Hz, 1 H, C5HBnth), 7.25 (d, 3J = 8.5 Hz, 2 H, C2/6HBenz), 6.92 (d, 3J = 8.5 Hz, 2 H, C3/5HBenz), 6.25 (d, 3J = 6.4 Hz, 1 H, C5HPyr), 5.21 (s, 2 H, N-CH2), 4.32 (s, 2 H, CH2-C=O) ppm. 13C NMR (DMSO-d6): δ = 167.4 (CH2-C=O), 162.1 (C2Pyr), 161.2 (C2Bnth), 160.9 (C4Pyr), 158.8 (C4Benz), 152.1 (C6HPyr), 151.4 (C3aBnth), 132.0 (C7aBnth), 128.8 (C2/6Benz), 126.8 (C1Benz), 124.6 (d, 1JCF = 271.8 Hz, CF3), 123.8 (d, 2JCF = 32.3 Hz, C6Bnth), 123.0 (d, 3JCF = 3.3 Hz, C5Bnth), 121.0 (C4Bnth), 120.0 (d, 3JCF = 4.2 Hz, C7Bnth), 114.0 (C3/5Benz), 110.2 (C5Pyr), 55.1 (OCH3), 46.5 (N-CH2), 36.0 (CH2-C=O) ppm. MS (ESI, 70 eV) m/z = 288.9 [C14H13N2O3S]+. IR: = 2936, 2849, 1649, 1560, 1497, 1373, 1319, 1256, 1244, 1159, 1111, 1084, 986, 827, 799, 677 cm−1.
2-((3-(4-Trifluoromethylbenzyl)-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoromethyl)benzo[d]thiazol-2-yl)acetamide (21): 2-((3-(4-Trifluoromethylbenzyl)-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoromethyl)benzo[d]thiazol-2-yl)acetamide (21) was obtained from 15 (105 mg, 0.36 mmol), 13 (97.5 mg, 0.34 mmol) and TEA (142 µL, 1.02 mmol). Purification by chromatography provided 21 as colorless solid. Yield: 71.1 mg (0.13 mmol, 38%). C22H14F6N4O2S2 (Mr 544.49). HPLC (purity): 100%. 1H NMR (DMSO-d6): δ = 12.94 (s, 1 H, NH), 8.50 (bs, 1 H, C7HBnth), 7.92 (d, 3J = 8.5 Hz, 1 H, C4HBnth), 7.83 (d, 3J = 6.5 Hz, 1 H, C6HPyr), 7.76–7.74 (m, 3 H, C5HBnth, C3/5HBenz), 7.49 (d, 3J = 8.1 Hz, 2 H, C2/6HBenz), 6.29 (d, 3J = 6.5 Hz, 1 H, C5HPyr), 5.38 (s, 2 H, N-CH2), 4.33 (s, 2 H, CH2-C=O) ppm. 13C NMR (DMSO-d6): δ = 167.2 (CH2-C=O), 162.1 (C2Pyr), 161.0 (C2Bnth), 160.9 (C4Pyr), 152.3 (C6HPyr), 151.3 (C3aBnth), 139.8 (C1Benz), 132.0 (C7aBnth), 128.2 (d, 2JCF = 31.8 Hz, C4Benz), 127.6 (C2/6Benz), 125.7 (d, 3JCF = 3.7 Hz, C3/5Benz), 124.6 (d, 1JCF = 272.5 Hz, CF3), 124.2 (d, 1JCF = 271.4 Hz, CF3-C4Benz), 123.9 (d, 2JCF = 32.1 Hz, C6Bnth), 123.1 (d, 3JCF = 3.1 Hz, C5Bnth), 121.1 (C4Bnth), 120.0 (d, 3JCF = 4.2 Hz, C7Bnth), 110.3 (C5Pyr), 46.7 (N-CH2), 35.9 (CH2-C=O) ppm. MS (ESI, 70 eV) m/z = 326.9 [C14H10F3N2O2S]+. IR: = 3065, 2978, 2922, 1655, 1562, 1493, 1319, 1153, 1119, 1111, 1066, 852, 799, 677, cm−1.
2-((3-(2-Fluoromethylbenzyl)-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoro-methyl)benzo[d]thiazol-2-yl)acetamide (22): 2-((3-(2-Fluoromethylbenzyl)-4-oxo-3,4-dihydropyrimidin-2-yl)thio)-N-(6-(trifluoro-methyl)benzo[d]thiazol-2-yl)acetamide (22) was obtained from 15 (67.7 mg, 0.23 mmol), 14 (48.5 mg, 0.21 mmol) and TEA (85.3 µL, 0.62 mmol). Purification by chromatography provided 22 as colorless solid. Yield: 82.2 mg (0.17 mmol, 81%). C21H14F4N4O2S2 (Mr 494.48). HPLC (purity): 100%. 1H NMR (DMSO-d6): δ = 12.94 (s, 1 H, NH), 8.50 (bs, 1 H, C7HBnth), 7.92 (d, 3J = 8.5 Hz, 1 H, C4HBnth), 7.83 (d, 3J = 6.5 Hz, 1 H, C6HPyr), 7.75 (dd, 3J = 8.6 Hz, 4J = 1.6 Hz, 1 H, C5HBnth), 7.41–7.35 (m, 1 H, C4HBenz), 7.30–7.25 (m, 1 H, C3HBenz), 7.19 (td, 3J = 7.5 Hz, 4J = 1.1 Hz, C5HBenz), 7.06–7.02 (m, 1 H, C6HBenz), 6.28 (d, 3J = 6.5 Hz, 1 H, C5HPyr), 5.31 (s, 2 H, N-CH2), 4.33 (s, 2 H, CH2-C=O) ppm. 13C NMR (DMSO-d6): δ = 167.2 (CH2-C=O), 162.1 (C2Pyr), 161.0 (C2Bnth), 160.8 (C4Pyr), 159.9 (d, 1JCF = 245.1 Hz, C2Benz), 152.3 (C6HPyr), 151.3 (C3aBnth), 132.0 (C7aBnth), 129.7 (d, 3JCF = 8.2 Hz, C4Benz), 127.5 (d, 3JCF = 3.5 Hz, C6Benz), 124.9 (d, 4JCF = 3.4 Hz, C5Benz), 124.6 (d, 1JCF = 271.8 Hz, CF3), 123.9 (d, 2JCF = 31.5 Hz, C6Bnth), 123.0 (d, 3JCF = 3.5 Hz, C5Bnth), 121.8 (d, 2JCF = 14.0 Hz, C1Benz), 121.1 (C4Bnth), 120.0 (d, 3JCF = 4.1 Hz, C7Bnth), 115.6 (d, 2JCF = 21.0 Hz, C3Benz), 110.2 (C5Pyr), 41.0 (d, 3JCF = 5.0 Hz, N-CH2), 35.8 (CH2-C=O) ppm. MS (ESI, 70 eV) m/z = 276.9 [C13H10FN2O2S]+. IR: = 3142, 3048, 2968, 2916, 1647, 1555, 1491, 1377, 1155, 1115, 1084, 826, 756, 679, cm−1.
4.10. Molecular Modeling
Molecular modeling was performed on a DELL Precision T5500 8-core system. For visualization Maestro version 11.1, Schrödinger LLC, (New York, NY, USA, 2017) was used. Protein crystal structures were prepared prior to docking by the Protein Preparation Wizard using default settings. The X-ray crystal structure refinement process included the addition of hydrogen atoms, optimization of hydrogen bonds, and removal of atomic clashes. Missing side chains and loops were filled in with Prime, and water molecules were deleted. Small molecule ligands were prepared to create energetically minimized 3D geometries as well as tautomer and ionization states using LigPrep. The ligand MgATP was energetically minimized using MacroModel. For ATP-docking, a homology model of CK1δ (PDB: 4twc) was created based on crystal structure PDB 1csn, a binary complex of CK1 with MgATP [
23]. Mutations were introduced into the model by using the biologics tool residue scanning and mutation with side-chain prediction and backbone minimization. Receptor grid generation was performed by Glide. For ligand docking, either the Glide XP workflow was used (default settings) or induced fit docking was used (glide setting: XP). For MgATP, induced fit docking automatic side-chain trimming was activated and H-bond constraints to the hinge binder residues Leu-85 and Glu-83 were specified. Energetically minimized small molecule ligand conformations were docked into the apo active site of the protein. Plausible binding poses were determined and subsequently ranked based on their calculated docking score.
4.11. Cell Lines and Transfection
Parental HeLa cells, as well as HeLa CSNK1D knock-out cells (CK1δ−/−) (generated by CRISP/Cas9 technology and provided by EDIGENE, Beijing, China), were cultured in Dulbecco’s Modified Eagle medium (DMEM). HCT-116 and HT-29 colon cancer cells were cultured in McCoy’s 5A medium whereas SW480 and SW620 colon cancer cells were cultured in Leibovitz’s L-15 medium. All media were supplemented with 10% (v/v) fetal bovine serum (FBS), 100 units/mL penicillin, and 100 µg/mL streptomycin. DMEM-based medium for the culture of HeLa CSNK1D knock-out cells was additionally supplemented with 1 μg/mL puromycin. Cell cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO2. Cells were transfected using Effectene transfection reagent (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. For selection of stably transfected clones, transfected cells were grown in selective medium, containing 0.6 mg/mL of G-418 (Calbiochem-Merck Millipore, Darmstadt, Germany).
4.12. MTT Viability Assay
For performing MTT viability assays, cells were seeded in a 96-well plate (5 × 104 cells/well for HeLa cells (parental, CK1δ−/−, or CK1δ−/− cells stably transfected with wild type or mutant CK1δ), HCT-116, and HT-29 cells or 7.5 × 104 cell/well for SW480 and SW620 cells). After 24 h, old medium was replaced with fresh medium containing either the small molecule of interest or DMSO as control. After 48 h of treatment, 10 μL MTT (5 mg/mL) was added and incubated at 37 °C for 4 h. The medium was then gently removed and 100 μL of acidic isopropanol was added (90% (v/v) isopropanol, 10% (v/v) 1 N HCl). Cell culture plates were then shaken for 30 min at room temperature. Absorbance was measured at 570 nm with TriStar2 LB942 Microplate Reader (Berthold Technologies, Bad Wildbad, Germany) and data analysis was performed using GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA).
4.13. Statistical Methods
Results were presented as the mean of experiments at least performed in triplicate ± SD. In the case of IC50 data, the standard error was calculated based on values obtained from three independent experiments. Enzyme kinetic data as well as IC50 and EC50 data was calculated using GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA). Other data calculation and presentation were performed using either GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA) or Microsoft Excel 2016 (Microsoft, Redmond, Washington, USA). Where indicated, statistical analysis was done by performing two-way ANOVA. P-values smaller than 0.05 were considered to be statistically significant. Levels of significance were indicated by asterisks as follows: * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001; ns, not significant.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}