Connexin 43 Mutations Lead to Increased Hemichannel Functionality in Skin Disease


Abstract
:1. Introduction
2. Connexins and the Epidermis
2.1. Gating Properties of Connexins
2.2. Connexins in Wound Healing
2.3. Cx43 Hemichannels
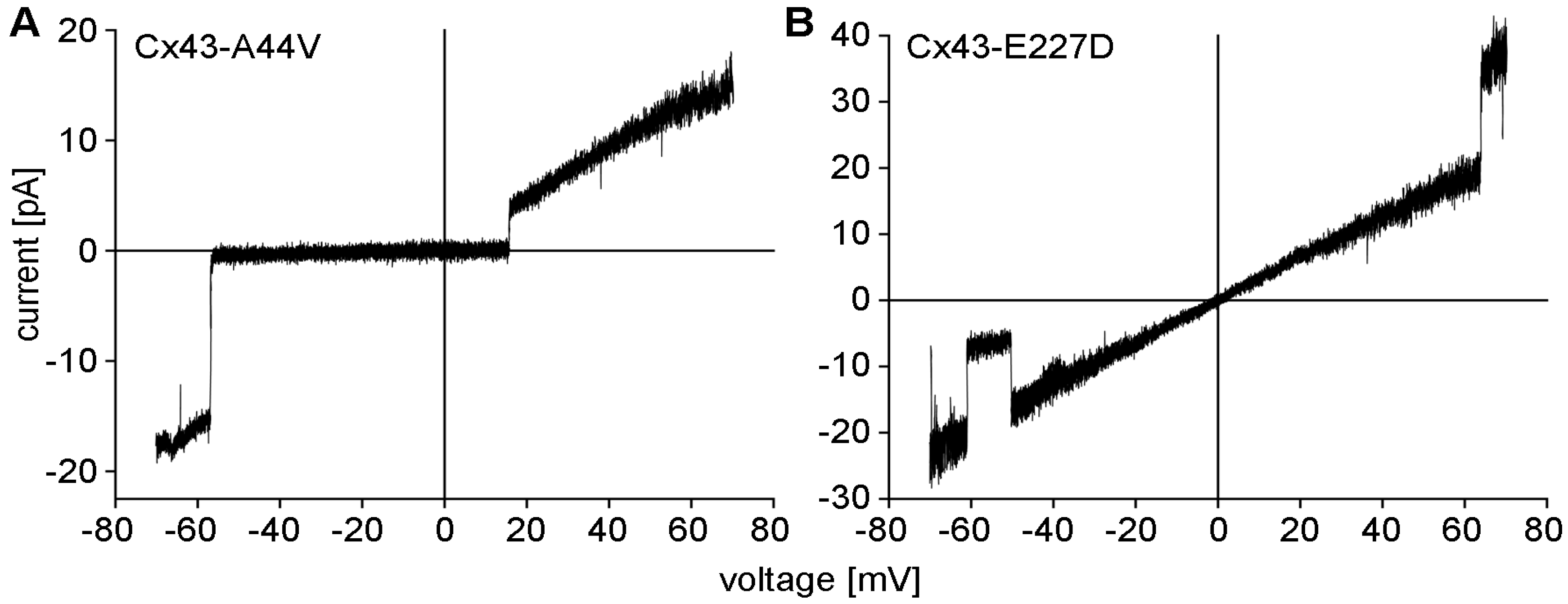
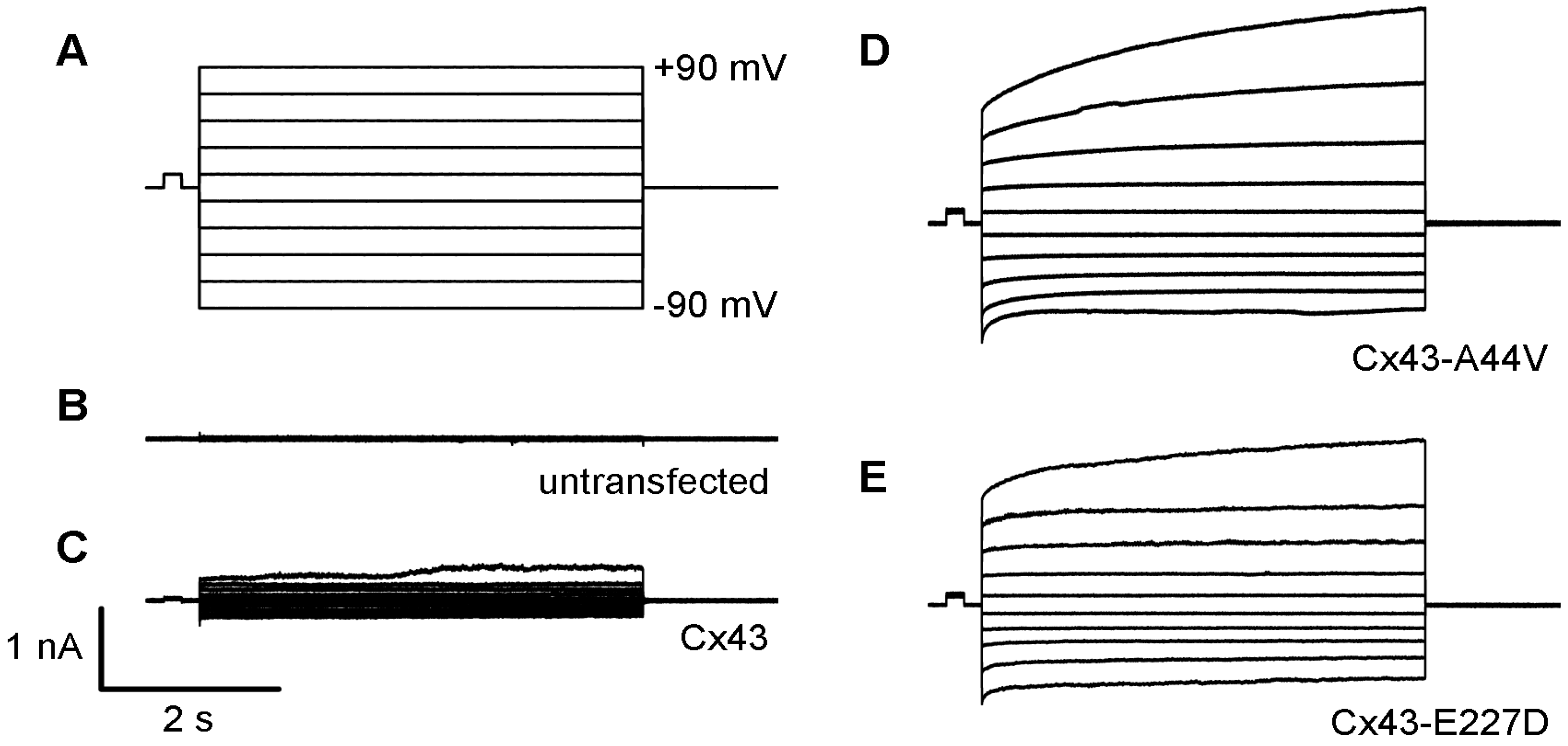
2.4. Cx43 Mutations and Epidermal Pathophysiology
2.5. A Generalizable Role for Hemichannel Activity?
2.6. Current Strategies for Skin Pathology
3. Conclusions
Funding
Conflicts of Interest
References
- Mese, G.; Richard, G.; White, T.W. Gap junctions: Basic structure and function. J. Investig. Dermatol. 2007, 127, 2516–2524. [Google Scholar] [CrossRef] [Green Version]
- Villanelo, F.; Escalona, Y.; Pareja-Barrueto, C.; Garate, J.A.; Skerrett, I.M.; Perez-Acle, T. Accessing gap-junction channel structure-function relationships through molecular modeling and simulations. BMC Cell Biol. 2017, 18, 5. [Google Scholar] [CrossRef] [Green Version]
- Herve, J.C.; Derangeon, M. Gap-junction-mediated cell-to-cell communication. Cell Tissue Res. 2013, 352, 21–31. [Google Scholar] [CrossRef]
- Wong, P.; Laxton, V.; Srivastava, S.; Chan, Y.W.; Tse, G. The role of gap junctions in inflammatory and neoplastic disorders (Review). Int. J. Mol. Med. 2017, 39, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Epifantseva, I.; Shaw, R.M. Intracellular trafficking pathways of Cx43 gap junction channels. Biochim. Biophys. Acta Biomembr. 2018, 1860, 40–47. [Google Scholar] [CrossRef]
- Nielsen, M.S.; Axelsen, L.N.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.H. Gap junctions. Compr. Physiol. 2012, 2, 1981–2035. [Google Scholar] [CrossRef] [Green Version]
- Kanaporis, G.; Mese, G.; Valiuniene, L.; White, T.W.; Brink, P.R.; Valiunas, V. Gap junction channels exhibit connexin-specific permeability to cyclic nucleotides. J. Gen. Physiol. 2008, 131, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, A.; Castro, C.; Flores, D.L.; Gutierrez, E.; Baldi, P. Gap Junction Channels of Innexins and Connexins: Relations and Computational Perspectives. Int. J. Mol. Sci. 2019, 20, 476. [Google Scholar] [CrossRef] [Green Version]
- Skerrett, I.M.; Williams, J.B. A structural and functional comparison of gap junction channels composed of connexins and innexins. Dev. Neurobiol. 2017, 77, 522–547. [Google Scholar] [CrossRef]
- Beyer, E.C.; Berthoud, V.M. The Family of Connexin Genes. Connexins 2009, 3–26. [Google Scholar] [CrossRef]
- Beyer, E.C.; Berthoud, V.M. Gap junction gene and protein families: Connexins, innexins, and pannexins. Biochim. Biophys. Acta Biomembr. 2018, 1860, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Söhl, G.; Willecke, K. An update on connexin genes and their nomenclature in mouse and man. Cell Commun. Adhes. 2003, 10, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Scheckenbach, K.E.; Crespin, S.; Kwak, B.R.; Chanson, M. Connexin channel-dependent signaling pathways in inflammation. J. Vasc. Res. 2011, 48, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Sohl, G.; Willecke, K. Gap junctions and the connexin protein family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, S.; Fiori, M.C.; Cuello, L.G.; Altenberg, G.A. A Cell-Based Assay to Assess Hemichannel Function. Yale J. Biol. Med. 2017, 90, 87–95. [Google Scholar]
- Willebrords, J.; Crespo Yanguas, S.; Maes, M.; Decrock, E.; Wang, N.; Leybaert, L.; Kwak, B.R.; Green, C.R.; Cogliati, B.; Vinken, M. Connexins and their channels in inflammation. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 413–439. [Google Scholar] [CrossRef]
- Osgood, R.S.; Upham, B.L.; Hill, T., 3rd; Helms, K.L.; Velmurugan, K.; Babica, P.; Bauer, A.K. Polycyclic aromatic hydrocarbon-induced signaling events relevant to inflammation and tumorigenesis in lung cells are dependent on molecular structure. PLoS ONE 2014, 8, e65150. [Google Scholar] [CrossRef] [Green Version]
- Upham, B.L.; Dwyer-Nield, L.D.; Bauer, A.K. Dysregulation of Gap Junction Function and Cytokine Production in Response to Non-Genotoxic Polycyclic Aromatic Hydrocarbons in an In Vitro Lung Cell Model. Cancers 2019, 11, 572. [Google Scholar] [CrossRef] [Green Version]
- Upham, B.L.; Blaha, L.; Babica, P.; Park, J.S.; Sovadinova, I.; Pudrith, C.; Rummel, A.M.; Weis, L.M.; Sai, K.; Tithof, P.K.; et al. Tumor promoting properties of a cigarette smoke prevalent polycyclic aromatic hydrocarbon as indicated by the inhibition of gap junctional intercellular communication via phosphatidylcholine-specific phospholipase C. Cancer Sci. 2008, 99, 696–705. [Google Scholar] [CrossRef] [Green Version]
- Osgood, R.S.; Upham, B.L.; Bushel, P.R.; Velmurugan, K.; Xiong, K.N.; Bauer, A.K. Secondhand Smoke-Prevalent Polycyclic Aromatic Hydrocarbon Binary Mixture-Induced Specific Mitogenic and Pro-inflammatory Cell Signaling Events in Lung Epithelial Cells. Toxicol. Sci. 2017, 157, 156–171. [Google Scholar] [CrossRef] [Green Version]
- Dosch, M.; Zindel, J.; Jebbawi, F.; Melin, N.; Sanchez-Taltavull, D.; Stroka, D.; Candinas, D.; Beldi, G. Connexin-43-dependent ATP release mediates macrophage activation during sepsis. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Masaki, K. Early disruption of glial communication via connexin gap junction in multiple sclerosis, Balo’s disease and neuromyelitis optica. Neuropathology 2015, 35, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Prakoura, N.; Kavvadas, P.; Chadjichristos, C.E. Connexin 43: A New Therapeutic Target Against Chronic Kidney Disease. Cell. Physiol. Biochem. 2018, 49, 985. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.E.; Li, L.; Xia, X.; Fu, W.; Liao, Q.; Lan, C.; Yang, D.; Chen, H.; Yue, R.; Zeng, C.; et al. Dedifferentiation, Proliferation, and Redifferentiation of Adult Mammalian Cardiomyocytes After Ischemic Injury. Circulation 2017, 136, 834–848. [Google Scholar] [CrossRef]
- George, S.A.; Poelzing, S. Cardiac conduction in isolated hearts of genetically modified mice—Connexin43 and salts. Prog. Biophys. Mol. Biol. 2016, 120, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Tien, T.; Muto, T.; Zhang, J.; Sohn, E.H.; Mullins, R.F.; Roy, S. Association of reduced Connexin 43 expression with retinal vascular lesions in human diabetic retinopathy. Exp. Eye. Res. 2016, 146, 103–106. [Google Scholar] [CrossRef]
- Nie, W.; Yan, H.; Li, S.; Zhu, W.; Fan, F.; Zhu, J. Angiotensin II Promotes Atherogenesis through upregulating the Expression of Connexin 43 in Dendritic Cells. Cell. Mol. Biol. Noisy Grand Fr. 2015, 61, 96–101. [Google Scholar]
- Tittarelli, A.; Guerrero, I.; Tempio, F.; Gleisner, M.A.; Avalos, I.; Sabanegh, S.; Ortiz, C.; Michea, L.; Lopez, M.N.; Mendoza-Naranjo, A.; et al. Overexpression of connexin 43 reduces melanoma proliferative and metastatic capacity. Br. J. Cancer 2015, 113, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Kretz, M.; Maass, K.; Willecke, K. Expression and function of connexins in the epidermis, analyzed with transgenic mouse mutants. Eur. J. Cell Biol. 2004, 83, 647–654. [Google Scholar] [CrossRef]
- Flenniken, A.M.; Osborne, L.R.; Anderson, N.; Ciliberti, N.; Fleming, C.; Gittens, J.E.; Gong, X.Q.; Kelsey, L.B.; Lounsbury, C.; Moreno, L.; et al. A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development 2005, 132, 4375–4386. [Google Scholar] [CrossRef] [Green Version]
- Lorentz, R.; Shao, Q.; Huang, T.; Fishman, G.I.; Laird, D.W. Characterization of gap junction proteins in the bladder of Cx43 mutant mouse models of oculodentodigital dysplasia. J. Membr. Biol. 2012, 245, 345–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, J.J.; Esseltine, J.L.; Shao, Q.; Jabs, E.W.; Sampson, J.; Auranen, M.; Bai, D.; Laird, D.W. Specific functional pathologies of Cx43 mutations associated with oculodentodigital dysplasia. Mol. Biol. Cell 2016, 27, 2172–2185. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W. Syndromic and non-syndromic disease-linked Cx43 mutations. FEBS Lett. 2014, 588, 1339–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, V.; Lau, A.F. Connexins: Mechanisms regulating protein levels and intercellular communication. FEBS Lett. 2014, 588, 1212–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivas, M.; Verselis, V.K.; White, T.W. Human diseases associated with connexin mutations. Biochim. Biophys. Acta Biomembr. 2018, 1860, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Reyes, E.P.; Garcia, I.E.; Pinto, B.; Martinez, A.D.; Gonzalez, C. Diseases associated with leaky hemichannels. Front. Cell. Neurosci. 2015, 9, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bargiello, T.A.; Oh, S.; Tang, Q.; Bargiello, N.K.; Dowd, T.L.; Kwon, T. Gating of Connexin Channels by transjunctional-voltage: Conformations and models of open and closed states. Biochim. Biophys. Acta Biomembr. 2018, 1860, 22–39. [Google Scholar] [CrossRef]
- Dosch, M.; Gerber, J.; Jebbawi, F.; Beldi, G. Mechanisms of ATP Release by Inflammatory Cells. Int. J. Mol. Sci. 2018, 19, 1222. [Google Scholar] [CrossRef] [Green Version]
- Bukauskas, F.F.; Jordan, K.; Bukauskiene, A.; Bennett, M.V.; Lampe, P.D.; Laird, D.W.; Verselis, V.K. Clustering of connexin 43–enhanced green fluorescent protein gap junction channels and functional coupling in living cells. Proc. Natl. Acad. Sci. USA 2000, 97, 2556–2561. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.X.; Gu, S. Gap junction-and hemichannel-independent actions of connexins. Biochim. Biophys. Acta BBA Biomembr. 2005, 1711, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Beyer, E.C.; Berthoud, V.M. Connexin hemichannels in the lens. Front. Physiol. 2014, 5, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasciani, I.; Temperan, A.; Perez-Atencio, L.F.; Escudero, A.; Martinez-Montero, P.; Molano, J.; Gomez-Hernandez, J.M.; Paino, C.L.; Gonzalez-Nieto, D.; Barrio, L.C. Regulation of connexin hemichannel activity by membrane potential and the extracellular calcium in health and disease. Neuropharmacology 2013, 75, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Valiunas, V.; Vogel, R.; Weingart, R. The kinetics of gap junction currents are sensitive to the ionic composition of the pipette solution. Pflug. Arch. 2000, 440, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Rossi, A.R.; Harris, A.L. Computational Studies of Molecular Permeation through Connexin26 Channels. Biophys. J. 2016, 110, 584–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valiunas, V.; Cohen, I.S.; Brink, P.R. Defining the factors that affect solute permeation of gap junction channels. Biochim. Biophys. Acta Biomembr. 2018, 1860, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Chanson, M.; Watanabe, M.; O’Shaughnessy, E.M.; Zoso, A.; Martin, P.E. Connexin Communication Compartments and Wound Repair in Epithelial Tissue. Int. J. Mol. Sci. 2018, 19, 1354. [Google Scholar] [CrossRef] [PubMed]
- Lilly, E.; Sellitto, C.; Milstone, L.M.; White, T.W. Connexin channels in congenital skin disorders. Semin. Cell Dev. Biol. 2016, 50, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Aasen, T. Connexins: Junctional and non-junctional modulators of proliferation. Cell Tissue Res. 2015, 360, 685–699. [Google Scholar] [CrossRef]
- Davis, N.G.; Phillips, A.; Becker, D.L. Connexin dynamics in the privileged wound healing of the buccal mucosa. Wound Repair Regen. 2013, 21, 571–578. [Google Scholar] [CrossRef]
- Cogliati, B.; Vinken, M.; Silva, T.C.; Araujo, C.M.M.; Aloia, T.P.A.; Chaible, L.M.; Mori, C.M.C.; Dagli, M.L.Z. Connexin 43 deficiency accelerates skin wound healing and extracellular matrix remodeling in mice. J. Dermatol. Sci. 2015, 79, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, J.; Ghatnekar, G.S.; Grek, C.L.; Moyer, K.E.; Gourdie, R.G. Connexin 43-Based Therapeutics for Dermal Wound Healing. Int. J. Mol. Sci. 2018, 19, 1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.; Nunan, R. Cellular and molecular mechanisms of repair in acute and chronic wound healing. Br. J. Dermatol. 2015, 173, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Cui, X. Connexin 43: Key roles in the skin. Biomed. Rep. 2017, 6, 605–611. [Google Scholar] [CrossRef] [Green Version]
- Lorraine, C.; Wright, C.S.; Martin, P.E. Connexin43 plays diverse roles in co-ordinating cell migration and wound closure events. Biochem. Soc. Trans. 2015, 43, 482–488. [Google Scholar] [CrossRef]
- Wright, C.S.; Van Steensel, M.A.; Hodgins, M.B.; Martin, P.E. Connexin mimetic peptides improve cell migration rates of human epidermal keratinocytes and dermal fibroblasts in vitro. Wound Repair Regen. 2009, 17, 240–249. [Google Scholar] [CrossRef]
- Xu, C.Y.; Zhang, W.S.; Zhang, H.; Cao, Y.; Zhou, H.Y. The Role of Connexin-43 in the Inflammatory Process: A New Potential Therapy to Influence Keratitis. J. Ophthalmol. 2019, 2019, 9312827. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Wang, X.; Qin, W.; Xu, X.; Xiong, Y.; Zhang, Y.; Zhang, H.; Sun, B. Endotoxin-induced autocrine ATP signaling inhibits neutrophil chemotaxis through enhancing myosin light chain phosphorylation. Proc. Natl. Acad. Sci. USA 2017, 114, 4483–4488. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Solan, J.L.; Lampe, P.D. Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim. Biophys. Acta Biomembr. 2018, 1860, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sosinsky, G.E.; Solan, J.L.; Gaietta, G.M.; Ngan, L.; Lee, G.J.; Mackey, M.R.; Lampe, P.D. The C-terminus of connexin43 adopts different conformations in the Golgi and gap junction as detected with structure-specific antibodies. Biochem. J. 2007, 408, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Ek-Vitorin, J.F.; Pontifex, T.K.; Burt, J.M. Cx43 Channel Gating and Permeation: Multiple Phosphorylation-Dependent Roles of the Carboxyl Terminus. Int. J. Mol. Sci. 2018, 19, 1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro-Rodrigues, T.M.; Martins-Marques, T.; Morel, S.; Kwak, B.R.; Girao, H. Role of connexin 43 in different forms of intercellular communication—Gap junctions, extracellular vesicles and tunnelling nanotubes. J. Cell Sci. 2017, 130, 3619–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, C.A.; Su, V.; Lau, A.F.; Lampe, P.D. Activation of Akt, not connexin 43 protein ubiquitination, regulates gap junction stability. J. Biol. Chem. 2012, 287, 2600–2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Guo, L.; Yang, X.; Wang, J.; Hou, Y.; Zhu, S.; Du, J.; Feng, J.; Xie, Y.; Zhuang, L.; et al. TGF-beta1-Induced Connexin43 Promotes Scar Formation via the Erk/MMP-1/Collagen III Pathway. J. Oral. Rehabil. 2019. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, G.T.; Lin, R.; Warn-Cramer, B.J.; Lau, A.F.; Burt, J.M. Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am. J. Physiol. Cell Physiol. 2003, 284, C511–C520. [Google Scholar] [CrossRef]
- Avshalumova, L.; Fabrikant, J.; Koriakos, A. Overview of Skin Diseases Linked to Connexin Gene Mutations. Int. J. Dermatol. 2014, 53, 192–205. [Google Scholar] [CrossRef]
- Srinivas, M.; Jannace, T.F.; Cocozzelli, A.G.; Li, L.; Slavi, N.; Sellitto, C.; White, T.W. Connexin43 mutations linked to skin disease have augmented hemichannel activity. Sci. Rep. 2019, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolski, R.; Sasse, P.; Schrickel, J.W.; Watkins, M.; Kim, J.S.; Rackauskas, M.; Troatz, C.; Ghanem, A.; Tiemann, K.; Degen, J.; et al. The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum. Mol. Genet. 2008, 17, 539–554. [Google Scholar] [CrossRef]
- Dobrowolski, R.; Sommershof, A.; Willecke, K. Some oculodentodigital dysplasia-associated Cx43 mutations cause increased hemichannel activity in addition to deficient gap junction channels. J. Membr. Biol. 2007, 219, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.Q.; Shao, Q.; Langlois, S.; Bai, D.; Laird, D.W. Differential potency of dominant negative connexin43 mutants in oculodentodigital dysplasia. J. Biol. Chem. 2007, 282, 19190–19202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida-Yamamoto, A. Erythrokeratodermia variabilis et progressiva. J. Dermatol. 2016, 43, 280–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Liang, J.; Chen, P.; Zeng, K.; Xue, R.; Tian, X.; Liang, L.; Wang, Q.; Shi, M.; Zhang, X. Two de novo GJA1 mutation in two sporadic patients with erythrokeratodermia variabilis et progressiva. Mol. Genet. Genomic. Med. 2019, 7, e670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Cao, X.; Lin, Z.; Lee, M.; Jia, X.; Ren, Y.; Dai, L.; Guan, L.; Zhang, J.; Lin, X.; et al. Exome sequencing reveals mutation in GJA1 as a cause of keratoderma-hypotrichosis-leukonychia totalis syndrome. Hum. Mol. Genet. 2015, 24, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Bursztejn, A.C.; Magdelaine, C.; Mortemousque, B.; Zerah, M.; Schmutz, J.L.; Leheup, B. Hypotrichosis with keratosis follicular and hyperostosis: A new phenotype due to GJA1 mutation. J. Eur. Acad. Dermatol. Venereol. 2019, 33, e219–e221. [Google Scholar] [CrossRef]
- Kelly, J.J.; Simek, J.; Laird, D.W. Mechanisms linking connexin mutations to human diseases. Cell Tissue Res. 2015, 360, 701–721. [Google Scholar] [CrossRef]
- Levit, N.A.; Mese, G.; Basaly, M.G.; White, T.W. Pathological hemichannels associated with human Cx26 mutations causing Keratitis-Ichthyosis-Deafness syndrome. Biochim. Biophys. Acta 2012, 1818, 2014–2019. [Google Scholar] [CrossRef] [Green Version]
- Garcia, I.E.; Maripillan, J.; Jara, O.; Ceriani, R.; Palacios-Munoz, A.; Ramachandran, J.; Olivero, P.; Perez-Acle, T.; Gonzalez, C.; Saez, J.C.; et al. Keratitis-ichthyosis-deafness syndrome-associated Cx26 mutants produce nonfunctional gap junctions but hyperactive hemichannels when co-expressed with wild type Cx43. J. Investig. Dermatol. 2015, 135, 1338–1347. [Google Scholar] [CrossRef] [Green Version]
- Shuja, Z.; Li, L.; Gupta, S.; Mese, G.; White, T.W. Connexin26 Mutations Causing Palmoplantar Keratoderma and Deafness Interact with Connexin43, Modifying Gap Junction and Hemichannel Properties. J. Investig. Dermatol. 2016, 136, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Basaran, E.; Yilmaz, E.; Alpsoy, E.; Yilmaz, G. Keratoderma, hypotrichosis and leukonychia totalis: A new syndrome? Br. J. Dermatol. 1995, 133, 636–638. [Google Scholar] [CrossRef] [PubMed]
- Boyden, L.M.; Craiglow, B.G.; Zhou, J.; Hu, R.; Loring, E.C.; Morel, K.D.; Lauren, C.T.; Lifton, R.P.; Bilguvar, K.; Paller, A.S.; et al. Dominant De Novo Mutations in GJA1 Cause Erythrokeratodermia Variabilis et Progressiva, without Features of Oculodentodigital Dysplasia. J. Investig. Dermatol. 2015, 135, 1540–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, G.; Brown, N.; Rouan, F.; Van der Schroeff, J.G.; Bijlsma, E.; Eichenfield, L.F.; Sybert, V.P.; Greer, K.E.; Hogan, P.; Campanelli, C.; et al. Genetic heterogeneity in erythrokeratodermia variabilis: Novel mutations in the connexin gene GJB4 (Cx30.3) and genotype-phenotype correlations. J. Investig. Dermatol. 2003, 120, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umegaki-Arao, N.; Sasaki, T.; Fujita, H.; Aoki, S.; Kameyama, K.; Amagai, M.; Seishima, M.; Kubo, A. Inflammatory Linear Verrucous Epidermal Nevus with a Postzygotic GJA1 Mutation Is a Mosaic Erythrokeratodermia Variabilis et Progressiva. J. Investig. Dermatol. 2017, 137, 967–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, J.; Mehregan, A.H. Inflammatory linear verrucose epidermal nevus. Arch. Dermatol. 1971, 104, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Essenfelder, G.M.; Bruzzone, R.; Lamartine, J.; Charollais, A.; Blanchet-Bardon, C.; Barbe, M.T.; Meda, P.; Waksman, G. Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activity. Hum. Mol. Genet. 2004, 13, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Li, L.; Liu, M.; Tan, J.; Tang, C.; Pan, Q.; Wang, D.; Zhang, Z. Pathogenic connexin-31 forms constitutively active hemichannels to promote necrotic cell death. PLoS ONE 2012, 7, e32531. [Google Scholar] [CrossRef] [Green Version]
- Maestrini, E.; Korge, B.P.; Ocana-Sierra, J.; Calzolari, E.; Cambiaghi, S.; Scudder, P.M.; Hovnanian, A.; Monaco, A.P.; Munro, C.S. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel’s syndrome) in three unrelated families. Hum. Mol. Genet. 1999, 8, 1237–1243. [Google Scholar] [CrossRef] [Green Version]
- Bakirtzis, G.; Jamieson, S.; Aasen, T.; Bryson, S.; Forrow, S.; Tetley, L.; Finbow, M.; Greenhalgh, D.; Hodgins, M. The effects of a mutant connexin 26 on epidermal differentiation. Cell Commun. Adhes. 2003, 10, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Becker, D.L.; Thrasivoulou, C.; Phillips, A.R. Connexins in wound healing; perspectives in diabetic patients. Biochim. Biophys. Acta 2012, 1818, 2068–2075. [Google Scholar] [CrossRef] [Green Version]
- Mori, R.; Power, K.T.; Wang, C.M.; Martin, P.; Becker, D.L. Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J. Cell Sci. 2006, 119, 5193–5203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretz, M.; Euwens, C.; Hombach, S.; Eckardt, D.; Teubner, B.; Traub, O.; Willecke, K.; Ott, T. Altered connexin expression and wound healing in the epidermis of connexin-deficient mice. J. Cell Sci. 2003, 116, 3443–3452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghatnekar, G.S.; Grek, C.L.; Armstrong, D.G.; Desai, S.C.; Gourdie, R.G. The effect of a connexin43-based Peptide on the healing of chronic venous leg ulcers: A multicenter, randomized trial. J. Investig. Dermatol. 2015, 135, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghatnekar, G.S.; O’Quinn, M.P.; Jourdan, L.J.; Gurjarpadhye, A.A.; Draughn, R.L.; Gourdie, R.G. Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen. Med. 2009, 4, 205–223. [Google Scholar] [CrossRef] [Green Version]
- Reaume, A.G.; de Sousa, P.A.; Kulkarni, S.; Langille, B.L.; Zhu, D.; Davies, T.C.; Juneja, S.C.; Kidder, G.M.; Rossant, J. Cardiac malformation in neonatal mice lacking connexin43. Science 1995, 267, 1831–1834. [Google Scholar] [CrossRef]
- Clarke, T.C.; Thomas, D.; Petersen, J.S.; Evans, W.H.; Martin, P.E. The antiarrhythmic peptide rotigaptide (ZP123) increases gap junction intercellular communication in cardiac myocytes and HeLa cells expressing connexin 43. Br. J. Pharmacol. 2006, 147, 486–495. [Google Scholar] [CrossRef] [Green Version]
- De Vuyst, E.; Boengler, K.; Antoons, G.; Sipido, K.R.; Schulz, R.; Leybaert, L. Pharmacological modulation of connexin-formed channels in cardiac pathophysiology. Br. J. Pharmacol. 2011, 163, 469–483. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Pathology | Connexin | Gene | Mutation | Clinical Features | Mechanism Linked to Pathology? |
|---|---|---|---|---|---|
| Erythrokeratoderma Variabilis et Progressiva (EKVP3) (OMIM 617525) | Cx43 | GJA1 | A44V (131C-T); E227D (681A-T); P283L (848C-T); T290N (869C-A) | Widespread or Localized Keratosis, Palmoplantar Keratoderma | Hemichannel Functionality or Unknown |
| Inflammatory Linear Verrucous Epidermal Nevus (ILVEN) | Cx43 | GJA1 | A44V (131C-T) | Raised Papules along Blaschko’s Lines | Hemichannel Functionality |
| Oculodentodigital Dysplasia (ODDD) (OMIM 164200) | Cx43 | GJA1 | Y17S (50A-C); S18P (52T-C); G21R (61G-A); G22E (65G-A); V96M (286G-A) 1 | Craniofacial, Dental, Ocular, and Digital Abnormalities, Syndactyly | Hemichannel Functionality |
| Palmoplantar Keratoderma and Congenital Alopecia-1 (PPKCA1) (OMIM 104100) | Cx43 | GJA1 | G8V (23G-T) | Keratosis of Palms, Knees, Elbows, and Feet, Alopecia, Leukonychia | Hemichannel Functionality |
| Hypotrichosis with Keratosis Follicular and Hyperostosis | Cx43 | GJA1 | G38E (113G-A) | Leukonychia, Palmoplantar Keratoderma, Hyperostosis, Alopecia | Unknown |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cocozzelli, A.G.; White, T.W. Connexin 43 Mutations Lead to Increased Hemichannel Functionality in Skin Disease. Int. J. Mol. Sci. 2019, 20, 6186. https://doi.org/10.3390/ijms20246186
Cocozzelli AG, White TW. Connexin 43 Mutations Lead to Increased Hemichannel Functionality in Skin Disease. International Journal of Molecular Sciences. 2019; 20(24):6186. https://doi.org/10.3390/ijms20246186
Chicago/Turabian StyleCocozzelli, Anthony G., and Thomas W. White. 2019. "Connexin 43 Mutations Lead to Increased Hemichannel Functionality in Skin Disease" International Journal of Molecular Sciences 20, no. 24: 6186. https://doi.org/10.3390/ijms20246186
APA StyleCocozzelli, A. G., & White, T. W. (2019). Connexin 43 Mutations Lead to Increased Hemichannel Functionality in Skin Disease. International Journal of Molecular Sciences, 20(24), 6186. https://doi.org/10.3390/ijms20246186