Gap Junctions in the Bone Marrow Lympho-Hematopoietic Stem Cell Niche, Leukemia Progression, and Chemoresistance


Abstract
:1. Introduction
2. Gap Junctions, Connexons, and Connexins
Connexin Biosynthesis and Turnover
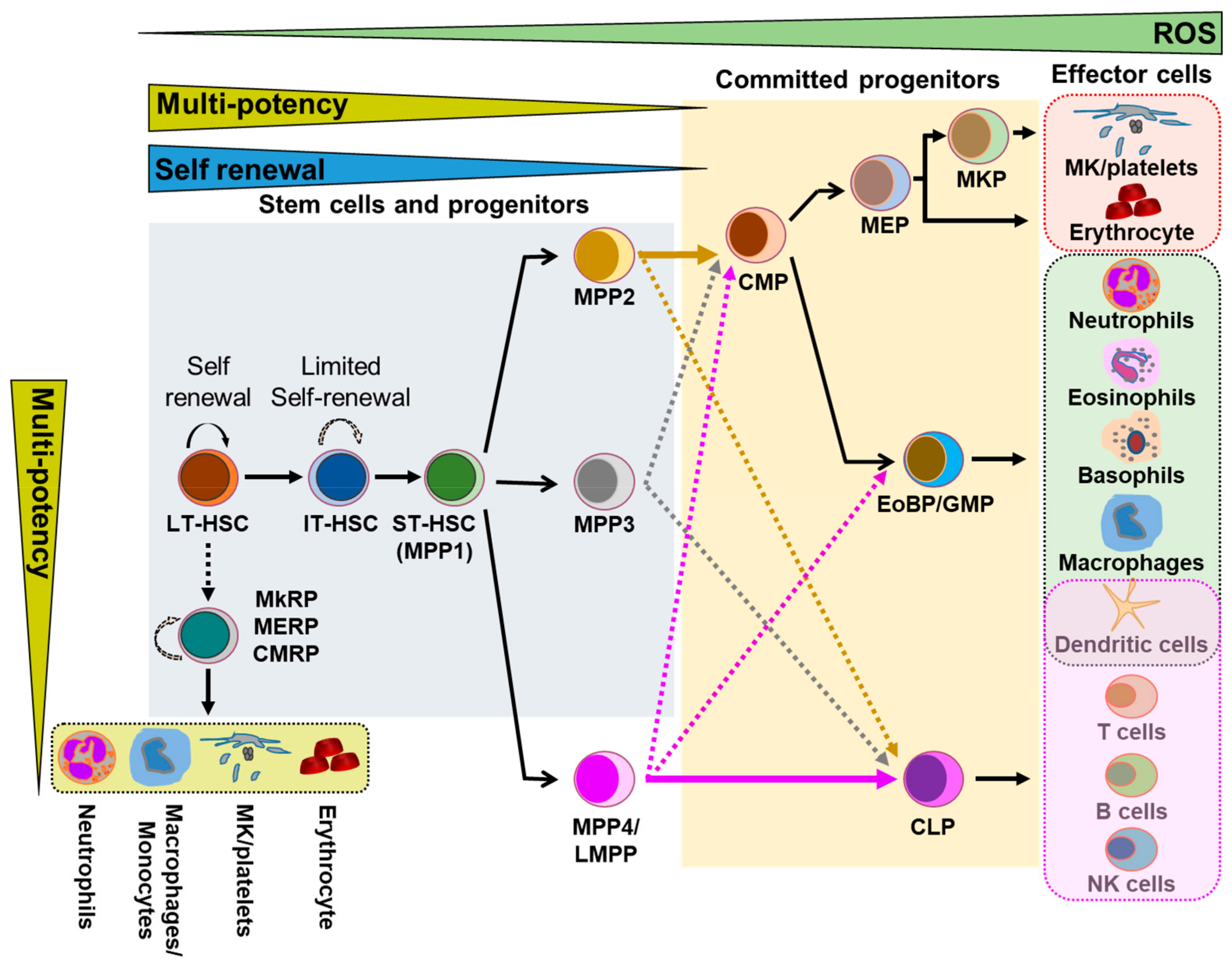
3. Involvement of GJIC in Hematopoiesis
3.1. Connexins in HSC
3.2. Gap Junctions in the HSC Niche
3.2.1. Osteoblasts
3.2.2. Mesenchymal Stem Cells
3.2.3. Endothelial Cells
3.2.4. Bone Marrow Macrophages
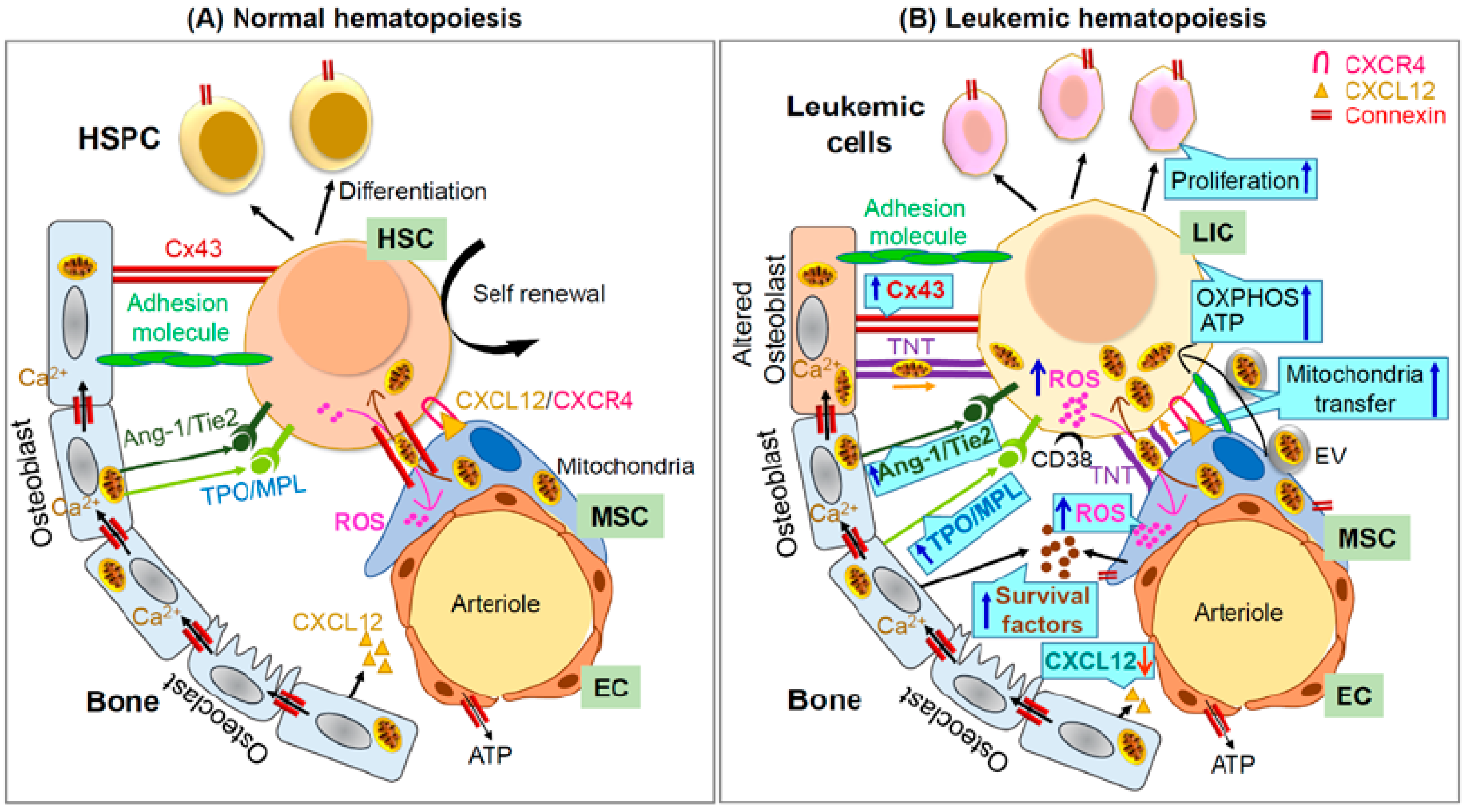
4. Role of Gap Junctions in Leukemic Hematopoiesis
Mitochondria Trafficking Mediated by Connexins in Leukemia
5. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
Abbreviations
| Atg16 | Autophagy related protein 16 |
| Akt | Protein kinase B |
| CCL4 | Chemokine C-C motif ligand 4 |
| CXCL12 | C-X-C motif chemokine 12 |
| CXCR | C-X-C chemokine receptor |
| ERK | Extracellular Signal-regulated Kinase |
| FAB | French-American-British classification |
| G-CSF | Granulocyte colony stimulating factor |
| GSK-3β | Gycogene Synthase Kinase-3-beta |
| IFNγ | Interferon gamma |
| IKK | Inhibitory kappa B kinase |
| IL-7 | Interleukin 7 |
| iPSC | Induced Pluripotent Stem Cells |
| LSK | Lin-/cKit+/Sca1+ |
| MEK | Mitogen-activated protein kinase kinase 1 |
| NADPH | Nicotine adenine dinucleotide phosphate, reduced form |
| NFκB | Nuclear factor kappa B |
| PDZ | domain shared by the postsynaptic density protein (PSD95), Drosophila disc large tumor suppressor (DlgA), and zonula occludens-1 protein (ZO-1) |
| Sp1 | Specificity protein 1 |
| STAT3 | Signal Transducer And Activator Of Transcription 3 |
| TNFα | Tumor Necrosis Factor alpha |
| TPO/MPL | Thrombopoietin/Myeloproliferative Leukemia protein (TPO receptor) |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
| Vps34 | Vacuolar Protein Sorting 34 |
References
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef]
- Jacobsen, S.E.W.; Nerlov, C. Haematopoiesis in the era of advanced single-cell technologies. Nat. Cell Biol. 2019, 21, 2–8. [Google Scholar] [CrossRef]
- Oguro, H.; Ding, L.; Morrison, S.J. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell. 2013, 13, 102–116. [Google Scholar] [CrossRef] [Green Version]
- Cabezas-Wallscheid, N.; Klimmeck, D.; Hansson, J.; Lipka, D.B.; Reyes, A.; Wang, Q.; Weichenhan, D.; Lier, A.; von Paleske, L.; Renders, S.; et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell. Stem Cell 2014, 15, 507–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitroulis, I.; Kalafati, L.; Hajishengallis, G.; Chavakis, T. Myelopoiesis in the Context of Innate Immunity. J. Innate. Immun. 2018, 10, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Nolan, D.J.; Vertes, E.L.; Varnum-Finney, B.; Kobayashi, H.; Hooper, A.T.; Seandel, M.; Shido, K.; White, I.A.; Kobayashi, M.; et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010, 6, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gori, J.L.; Butler, J.M.; Kunar, B.; Poulos, M.G.; Ginsberg, M.; Nolan, D.J.; Norgaard, Z.K.; Adair, J.E.; Rafii, S.; Kiem, H.P. Endothelial Cells Promote Expansion of Long-Term Engrafting Marrow Hematopoietic Stem and Progenitor Cells in Primates. Stem Cell. Transl. Med. 2017, 6, 864–876. [Google Scholar] [CrossRef]
- Schajnovitz, A.; Itkin, T.; D’Uva, G.; Kalinkovich, A.; Golan, K.; Ludin, A.; Cohen, D.; Shulman, Z.; Avigdor, A.; Nagler, A.; et al. CXCL12 secretion by bone marrow stromal cells is dependent on cell contact and mediated by connexin-43 and connexin-45 gap junctions. Nat. Immunol. 2011, 12, 391–398. [Google Scholar] [CrossRef]
- Chang, K.H.; Nayak, R.C.; Roy, S.; Perumbeti, A.; Wellendorf, A.M.; Bezold, K.Y.; Pirman, M.; Hill, S.E.; Starnes, J.; Loberg, A.; et al. Vasculopathy-associated hyperangiotensinemia mobilizes haematopoietic stem cells/progenitors through endothelial AT(2)R and cytoskeletal dysregulation. Nat. Commun. 2015, 6, 5914. [Google Scholar] [CrossRef] [Green Version]
- Abascal, F.; Zardoya, R. Evolutionary analyses of gap junction protein families. Biochim. Biophys. Acta 2013, 1828, 4–14. [Google Scholar] [CrossRef]
- Laird, D.W.; Lampe, P.D. Therapeutic strategies targeting connexins. Nat. Rev. Drug Discov. 2018, 17, 905–921. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Nieto, D.; Chang, K.H.; Fasciani, I.; Nayak, R.; Fernandez-Garcia, L.; Barrio, L.C.; Cancelas, J.A. Connexins: Intercellular Signal Transmitters in Lymphohematopoietic Tissues. Int. Rev. Cell Mol. Biol. 2015, 318, 27–62. [Google Scholar] [PubMed]
- Sohl, G.; Willecke, K. Gap junctions and the connexin protein family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Montecino-Rodriguez, E.; Leathers, H.; Dorshkind, K. Expression of connexin 43 (Cx43) is critical for normal hematopoiesis. Blood 2000, 96, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi Ishikawa, E.; Gonzalez-Nieto, D.; Ghiaur, G.; Dunn, S.K.; Ficker, A.M.; Murali, B.; Madhu, M.; Gutstein, D.E.; Fishman, G.I.; Barrio, L.C.; et al. Connexin-43 prevents hematopoietic stem cell senescence through transfer of reactive oxygen species to bone marrow stromal cells. Proc. Natl. Acad. Sci. USA 2012, 109, 9071–9076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presley, C.A.; Lee, A.W.; Kastl, B.; Igbinosa, I.; Yamada, Y.; Fishman, G.I.; Gutstein, D.E.; Cancelas, J.A. Bone marrow connexin-43 expression is critical for hematopoietic regeneration after chemotherapy. Cell Commun. Adhes. 2005, 12, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancelas, J.A.; Koevoet, W.L.; de Koning, A.E.; Mayen, A.E.; Rombouts, E.J.; Ploemacher, R.E. Connexin-43 gap junctions are involved in multiconnexin-expressing stromal support of hemopoietic progenitors and stem cells. Blood 2000, 96, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Rosendaal, M.; Green, C.R.; Rahman, A.; Morgan, D. Up-regulation of the connexin43+ gap junction network in haemopoietic tissue before the growth of stem cells. J. Cell Sci. 1994, 107, 29–37. [Google Scholar]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef] [Green Version]
- Miraki-Moud, F.; Anjos-Afonso, F.; Hodby, K.A.; Griessinger, E.; Rosignoli, G.; Lillington, D.; Jia, L.; Davies, J.K.; Cavenagh, J.; Smith, M.; et al. Acute myeloid leukemia does not deplete normal hematopoietic stem cells but induces cytopenias by impeding their differentiation. Proc. Natl. Acad. Sci. USA 2013, 110, 13576–13581. [Google Scholar] [CrossRef] [Green Version]
- Suganuma, K.; Miwa, H.; Imai, N.; Shikami, M.; Gotou, M.; Goto, M.; Mizuno, S.; Takahashi, M.; Yamamoto, H.; Hiramatsu, A.; et al. Energy metabolism of leukemia cells: Glycolysis versus oxidative phosphorylation. Leuk Lymphoma 2010, 51, 2112–2119. [Google Scholar] [CrossRef] [PubMed]
- Ciciarello, M.; Corradi, G.; Loscocco, F.; Visani, G.; Monaco, F.; Cavo, M.; Curti, A.; Isidori, A. The Yin and Yang of the Bone Marrow Microenvironment: Pros and Cons of Mesenchymal Stromal Cells in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samudio, I.; Fiegl, M.; McQueen, T.; Clise-Dwyer, K.; Andreeff, M. The warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res. 2008, 68, 5198–5205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopleva, M.; Konoplev, S.; Hu, W.; Zaritskey, A.Y.; Afanasiev, B.V.; Andreeff, M. Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia 2002, 16, 1713–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodenough, D.A.; Goliger, J.A.; Paul, D.L. Connexins, connexons, and intercellular communication. Annu. Rev. Biochem. 1996, 65, 475–502. [Google Scholar] [CrossRef] [PubMed]
- White, T.W.; Bruzzone, R. Multiple connexin proteins in single intercellular channels: Connexin compatibility and functional consequences. J. Bioenerg. Biomembr. 1996, 28, 339–350. [Google Scholar] [CrossRef]
- Beyer, E.C.; Davis, L.M.; Saffitz, J.E.; Veenstra, R.D. Cardiac intercellular communication: Consequences of connexin distribution and diversity. Braz. J. Med. Biol. Res. 1995, 28, 415–425. [Google Scholar]
- Forsberg, E.C.; Prohaska, S.S.; Katzman, S.; Heffner, G.C.; Stuart, J.M.; Weissman, I.L. Differential expression of novel potential regulators in hematopoietic stem cells. PLoS Genet. 2005, 1, e28. [Google Scholar] [CrossRef] [Green Version]
- Hirabayashi, Y.; Yoon, B.I.; Tsuboi, I.; Huo, Y.; Kodama, Y.; Kanno, J.; Ott, T.; Trosko, J.E.; Inoue, T. Membrane channel connexin 32 maintains Lin(-)/c-kit(+) hematopoietic progenitor cell compartment: Analysis of the cell cycle. J. Membr. Biol. 2007, 217, 105–113. [Google Scholar] [CrossRef]
- Alves, L.A.; Campos de Carvalho, A.C.; Cirne Lima, E.O.; Rocha e Souza, C.M.; Dardenne, M.; Spray, D.C.; Savino, W. Functional gap junctions in thymic epithelial cells are formed by connexin 43. Eur. J. Immunol. 1995, 25, 431–437. [Google Scholar] [CrossRef]
- Alves, L.A.; Coutinho-Silva, R.; Persechini, P.M.; Spray, D.C.; Savino, W.; Campos de Carvalho, A.C. Are there functional gap junctions or junctional hemichannels in macrophages? Blood 1996, 88, 328–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado, S.P.; Balduino, A.; Bodi, E.C.; El-Cheikh, M.C.; Campos de Carvalho, A.C.; Borojevic, R. Connexin expression and gap-junction-mediated cell interactions in an in vitro model of haemopoietic stroma. Cell. Tissue Res. 2004, 316, 65–76. [Google Scholar] [CrossRef]
- Berthoud, V.M.; Minogue, P.J.; Laing, J.G.; Beyer, E.C. Pathways for degradation of connexins and gap junctions. Cardiovasc. Res. 2004, 62, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.X.; Paul, D.L.; Goodenough, D.A. Posttranslational phosphorylation of lens fiber connexin46: A slow occurrence. Invest. Ophthalmol. Vis. Sci. 1993, 34, 3558–3565. [Google Scholar]
- Kelly, J.J.; Shao, Q.; Jagger, D.J.; Laird, D.W. Cx30 exhibits unique characteristics including a long half-life when assembled into gap junctions. J. Cell Sci. 2015, 128, 3947–3960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koval, M. Pathways and control of connexin oligomerization. Trends Cell Biol. 2006, 16, 159–166. [Google Scholar]
- Koval, M.; Molina, S.A.; Burt, J.M. Mix and match: Investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 2014, 588, 1193–1204. [Google Scholar] [CrossRef] [Green Version]
- John, S.A.; Revel, J.P. Connexon integrity is maintained by non-covalent bonds: Intramolecular disulfide bonds link the extracellular domains in rat connexin-43. Biochem. Biophys. Res. Commun. 1991, 178, 1312–1318. [Google Scholar] [CrossRef]
- Musil, L.S.; Goodenough, D.A. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell 1993, 74, 1065–1077. [Google Scholar] [CrossRef]
- Giepmans, B.N.; Verlaan, I.; Hengeveld, T.; Janssen, H.; Calafat, J.; Falk, M.M.; Moolenaar, W.H. Gap junction protein connexin-43 interacts directly with microtubules. Curr. Biol. 2001, 11, 1364–1368. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.M.; Fay, A.J.; Puthenveedu, M.A.; von Zastrow, M.; Jan, Y.N.; Jan, L.Y. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 2007, 128, 547–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaietta, G.; Deerinck, T.J.; Adams, S.R.; Bouwer, J.; Tour, O.; Laird, D.W.; Sosinsky, G.E.; Tsien, R.Y.; Ellisman, M.H. Multicolor and electron microscopic imaging of connexin trafficking. Science 2002, 296, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Lauf, U.; Giepmans, B.N.; Lopez, P.; Braconnot, S.; Chen, S.C.; Falk, M.M. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 10446–10451. [Google Scholar] [CrossRef] [Green Version]
- Jordan, K.; Chodock, R.; Hand, A.R.; Laird, D.W. The origin of annular junctions: A mechanism of gap junction internalization. J. Cell Sci. 2001, 114, 763–773. [Google Scholar] [PubMed]
- Laird, D.W. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim. Biophys. Acta 2005, 1711, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laing, J.G.; Manley-Markowski, R.N.; Koval, M.; Civitelli, R.; Steinberg, T.H. Connexin45 interacts with zonula occludens-1 and connexin43 in osteoblastic cells. J. Biol. Chem. 2001, 276, 23051–23055. [Google Scholar] [CrossRef] [Green Version]
- Giepmans, B.N.; Verlaan, I.; Moolenaar, W.H. Connexin-43 interactions with ZO-1 and alpha- and beta-tubulin. Cell Commun. Adhes. 2001, 8, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Hunter, A.W.; Jourdan, J.; Gourdie, R.G. Fusion of GFP to the carboxyl terminus of connexin43 increases gap junction size in HeLa cells. Cell Commun. Adhes. 2003, 10, 211–214. [Google Scholar] [CrossRef]
- Levy, J.A.; Weiss, R.M.; Dirksen, E.R.; Rosen, M.R. Possible communication between murine macrophages oriented in linear chains in tissue culture. Exp. Cell Res. 1976, 103, 375–385. [Google Scholar] [CrossRef]
- Porvaznik, M.; MacVittie, T.J. Detection of gap junctions between the progeny of a canine macrophage colony-forming cell in vitro. J. Cell Biol. 1979, 82, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Kapsenberg, M.L.; Leene, W. Formation of B type gap junctions between PHA-stimulated rabbit lymphocytes. Exp. Cell Res. 1979, 120, 211–222. [Google Scholar] [CrossRef]
- Neumark, T.; Huynh, D.C. Gap junctions between human T-colony cells. Acta Morphol. Hung. 1989, 37, 147–153. [Google Scholar] [PubMed]
- Dorshkind, K.; Green, L.; Godwin, A.; Fletcher, W.H. Connexin-43-type gap junctions mediate communication between bone marrow stromal cells. Blood 1993, 82, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Campbell, F.R. Gap junctions between cells of bone marrow: An ultrastructural study using tannic acid. Anat. Rec. 1980, 196, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K. Sl/Sld mice have an increased number of gap junctions in their bone marrow stromal cells. Blood Cell. 1988, 13, 421–435. [Google Scholar]
- Yamazaki, K.; Allen, T.D. Ultrastructural morphometric study of efferent nerve terminals on murine bone marrow stromal cells, and the recognition of a novel anatomical unit: The “neuro-reticular complex”. Am. J. Anat. 1990, 187, 261–276. [Google Scholar] [CrossRef]
- Yamazaki, K.; Allen, T.D. Ultrastructural and morphometric alterations in bone marrow stromal tissue after 7 Gy irradiation. Blood Cell. 1991, 17, 527–549. [Google Scholar]
- Yamazaki, K.; Eyden, B.P. A study of intercellular relationships between trabecular bone and marrow stromal cells in the murine femoral metaphysis. Anat. Embryol. 1995, 192, 9–20. [Google Scholar] [CrossRef]
- Yamazaki, K.; Zacharov, Y.; Simmons, P.J.; Dexter, T.M.; Allen, T.D. A comparative morphometric study on the ultrastructure of adherent cells in long-term bone marrow culture from normal and congenitally anemic mice. Blood Cell. 1989, 15, 343–364. [Google Scholar]
- Krenacs, T.; Rosendaal, M. Immunohistological detection of gap junctions in human lymphoid tissue: connexin43 in follicular dendritic and lymphoendothelial cells. J. Histochem. Cytochem. 1995, 43, 1125–1137. [Google Scholar] [CrossRef] [Green Version]
- Krenacs, T.; Rosendaal, M. Gap-junction communication pathways in germinal center reactions. Dev. Immunol. 1998, 6, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Krenacs, T.; Rosendaal, M. Connexin43 gap junctions in normal, regenerating, and cultured mouse bone marrow and in human leukemias: Their possible involvement in blood formation. Am. J. Pathol. 1998, 152, 993–1004. [Google Scholar] [PubMed]
- Krenacs, T.; van Dartel, M.; Lindhout, E.; Rosendaal, M. Direct cell/cell communication in the lymphoid germinal center: connexin43 gap junctions functionally couple follicular dendritic cells to each other and to B lymphocytes. Eur. J. Immunol. 1997, 27, 1489–1497. [Google Scholar] [CrossRef]
- Rosendaal, M. Gap junctions in blood forming tissues. Microsc. Res. Tech. 1995, 31, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Rosendaal, M.; Gregan, A.; Green, C.R. Direct cell-cell communication in the blood-forming system. Tis Cell. 1991, 23, 457–470. [Google Scholar] [CrossRef]
- Rosendaal, M.; Jopling, C. Hematopoietic capacity of connexin43 wild-type and knock-out fetal liver cells not different on wild-type stroma. Blood 2003, 101, 2996–2998. [Google Scholar] [CrossRef] [Green Version]
- Rosendaal, M.; Krenacs, T.T. Regulatory pathways in blood-forming tissue with particular reference to gap junctional communication. Pathol. Oncol. Res. 2000, 6, 243–249. [Google Scholar] [CrossRef]
- Rosendaal, M.; Mayen, A.; de Koning, A.; Dunina-Barkovskaya, T.; Krenacs, T.; Ploemacher, R. Does transmembrane communication through gap junctions enable stem cells to overcome stromal inhibition? Leukemia 1997, 11, 1281–1289. [Google Scholar] [CrossRef] [Green Version]
- Rosendaal, M.; Stone, M. Enhancement of repopulation haemopoiesis by heterozygous connexin 43 stem cells seeded on wild-type connexin 43 stroma. Clin. Sci. 2003, 105, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Bermudez-Fajardo, A.; Yliharsila, M.; Evans, W.H.; Newby, A.C.; Oviedo-Orta, E. CD4+ T lymphocyte subsets express connexin 43 and establish gap junction channel communication with macrophages in vitro. J. Leukoc. Biol. 2007, 82, 608–612. [Google Scholar] [CrossRef]
- Oviedo-Orta, E.; Errington, R.J.; Evans, W.H. Gap junction intercellular communication during lymphocyte transendothelial migration. Cell. Biol. Int. 2002, 26, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Oviedo-Orta, E.; Evans, W.H. Gap junctions and connexins: Potential contributors to the immunological synapse. J. Leukoc. Biol. 2002, 72, 636–642. [Google Scholar] [PubMed]
- Oviedo-Orta, E.; Gasque, P.; Evans, W.H. Immunoglobulin and cytokine expression in mixed lymphocyte cultures is reduced by disruption of gap junction intercellular communication. FASEB J. 2001, 15, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Oviedo-Orta, E.; Hoy, T.; Evans, W.H. Intercellular communication in the immune system: Differential expression of connexin40 and 43, and perturbation of gap junction channel functions in peripheral blood and tonsil human lymphocyte subpopulations. Immunology 2000, 99, 578–590. [Google Scholar] [CrossRef]
- Oviedo-Orta, E.; Perreau, M.; Evans, W.H.; Potolicchio, I. Control of the proliferation of activated CD4+ T cells by connexins. J. Leukoc. Biol. 2010, 88, 79–86. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Yoon, B.I.; Tsuboi, I.; Huo, Y.; Kodama, Y.; Kanno, J.; Ott, T.; Trosko, J.E.; Inoue, T. Protective role of connexin 32 in steady-state hematopoiesis, regeneration state, and leukemogenesis. Exp. Biol. Med. 2007, 232, 700–712. [Google Scholar]
- Liu, Y.; Elf, S.E.; Miyata, Y.; Sashida, G.; Liu, Y.; Huang, G.; Di Giandomenico, S.; Lee, J.M.; Deblasio, A.; Menendez, S.; et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 2009, 4, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef]
- Singh, A.K.; Althoff, M.J.; Cancelas, J.A. Signaling Pathways Regulating Hematopoietic Stem Cell and Progenitor Aging. Curr. Stem Cell Rep. 2018, 4, 166–181. [Google Scholar] [CrossRef]
- Durig, J.; Rosenthal, C.; Halfmeyer, K.; Wiemann, M.; Novotny, J.; Bingmann, D.; Duhrsen, U.; Schirrmacher, K. Intercellular communication between bone marrow stromal cells and CD34+ haematopoietic progenitor cells is mediated by connexin 43-type gap junctions. Br. J. Haematol. 2000, 111, 416–425. [Google Scholar] [CrossRef]
- Bejarano, E.; Yuste, A.; Patel, B.; Stout, R.F., Jr.; Spray, D.C.; Cuervo, A.M. Connexins modulate autophagosome biogenesis. Nat. Cell Biol. 2014, 16, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.N.; Kwon, H.J.; Im, S.W.; Son, Y.H.; Akindehin, S.; Jung, Y.S.; Lee, S.J.; Rhyu, I.J.; Kim, I.Y.; Seong, J.K.; et al. Connexin 43 is required for the maintenance of mitochondrial integrity in brown adipose tissue. Sci. Rep. 2017, 7, 7159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannini, N.; Campos, V.; Girotra, M.; Trachsel, V.; Rojas-Sutterlin, S.; Tratwal, J.; Ragusa, S.; Stefanidis, E.; Ryu, D.; Rainer, P.Y.; et al. The NAD-Booster Nicotinamide Riboside Potently Stimulates Hematopoiesis through Increased Mitochondrial Clearance. Cell Stem Cell. 2019, 24, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Turcotte, R.; Cui, J.; Zimmerman, S.E.; Pinho, S.; Mizoguchi, T.; Arai, F.; Runnels, J.M.; Alt, C.; Teruya-Feldstein, J.; et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 2016, 354, 1156–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegue, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Golan, K.; Althoff, M.J.; Petrovich-Kopitman, E.; Wellendorf, A.M.; Mohmoud, F.; Bertagna, M.; Gonzalez-Nieto, D.; Barrio, L.C.; Lapidot, T.; et al. Connexin-43 Is a Negative Regulator of Mitochondrial Fission, Mitophagy and Apoptosis of Dividing Hematopoietic Stem Cells through the Drp1-Pink1 Axis. Blood 2018, 132, 639. [Google Scholar] [CrossRef]
- Civitelli, R. Cell-cell communication in the osteoblast/osteocyte lineage. Arch. Biochem. Biophys. 2008, 473, 188–192. [Google Scholar] [CrossRef] [Green Version]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Civitelli, R.; Beyer, E.C.; Warlow, P.M.; Robertson, A.J.; Geist, S.T.; Steinberg, T.H. Connexin43 mediates direct intercellular communication in human osteoblastic cell networks. J. Clin. Invest. 1993, 91, 1888–1896. [Google Scholar] [CrossRef] [Green Version]
- Wagner, W.; Wein, F.; Roderburg, C.; Saffrich, R.; Diehlmann, A.; Eckstein, V.; Ho, A.D. Adhesion of human hematopoietic progenitor cells to mesenchymal stromal cells involves CD44. Cell. Tissues Org. 2008, 188, 160–169. [Google Scholar] [CrossRef]
- Oviedo-Orta, E.; Evans, W.H. Gap junctions and connexin-mediated communication in the immune system. Biochim. Biophys. Acta 2004, 1662, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Guillotin, B.; Bourget, C.; Remy-Zolgadri, M.; Bareille, R.; Fernandez, P.; Conrad, V.; Amedee-Vilamitjana, J. Human primary endothelial cells stimulate human osteoprogenitor cell differentiation. Cell Physiol. Biochem. 2004, 14, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Ploemacher, R.E.; Mayen, A.E.; De Koning, A.E.; Krenacs, T.; Rosendaal, M. Hematopoiesis: Gap Junction Intercellular Communication is Likely to be Involved in Regulation of Stroma-dependent Proliferation of Hemopoietic Stem Cells. Hematology 2000, 5, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573–590. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Reilly, M.J.; Emerson, S.G. Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood 1996, 87, 518–524. [Google Scholar] [CrossRef]
- Taichman, R.; Reilly, M.; Verma, R.; Ehrenman, K.; Emerson, S. Hepatocyte growth factor is secreted by osteoblasts and cooperatively permits the survival of haematopoietic progenitors. Br. J. Haematol. 2001, 112, 438–448. [Google Scholar] [CrossRef]
- El-Badri, N.S.; Wang, B.Y.; Good, R.A. Osteoblasts promote engraftment of allogeneic hematopoietic stem cells. Exp. Hematol. 1998, 26, 110–116. [Google Scholar]
- Nilsson, S.K.; Johnston, H.M.; Whitty, G.A.; Williams, B.; Webb, R.J.; Denhardt, D.T.; Bertoncello, I.; Bendall, L.J.; Simmons, P.J.; Haylock, D.N. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005, 106, 1232–1239. [Google Scholar] [CrossRef]
- Stier, S.; Ko, Y.; Forkert, R.; Lutz, C.; Neuhaus, T.; Grunewald, E.; Cheng, T.; Dombkowski, D.; Calvi, L.M.; Rittling, S.R.; et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J. Exp. Med. 2005, 201, 1781–1791. [Google Scholar] [CrossRef]
- Arai, F.; Hirao, A.; Ohmura, M.; Sato, H.; Matsuoka, S.; Takubo, K.; Ito, K.; Koh, G.Y.; Suda, T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 2004, 118, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulyanova, T.; Scott, L.M.; Priestley, G.V.; Jiang, Y.; Nakamoto, B.; Koni, P.A.; Papayannopoulou, T. VCAM-1 expression in adult hematopoietic and nonhematopoietic cells is controlled by tissue-inductive signals and reflects their developmental origin. Blood 2005, 106, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Visnjic, D.; Kalajzic, Z.; Rowe, D.W.; Katavic, V.; Lorenzo, J.; Aguila, H.L. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 2004, 103, 3258–3264. [Google Scholar] [CrossRef] [PubMed]
- Loiselle, A.E.; Paul, E.M.; Lewis, G.S.; Donahue, H.J. Osteoblast and osteocyte-specific loss of Connexin43 results in delayed bone formation and healing during murine fracture healing. J. Orthop. Res. 2013, 31, 147–154. [Google Scholar] [CrossRef]
- Loiselle, A.E.; Lloyd, S.A.; Paul, E.M.; Lewis, G.S.; Donahue, H.J. Inhibition of GSK-3beta rescues the impairments in bone formation and mechanical properties associated with fracture healing in osteoblast selective connexin 43 deficient mice. PLoS ONE 2013, 8, e81399. [Google Scholar] [CrossRef] [Green Version]
- Bivi, N.; Condon, K.W.; Allen, M.R.; Farlow, N.; Passeri, G.; Brun, L.R.; Rhee, Y.; Bellido, T.; Plotkin, L.I. Cell autonomous requirement of connexin 43 for osteocyte survival: Consequences for endocortical resorption and periosteal bone formation. J. Bone Miner. Res. 2012, 27, 374–389. [Google Scholar] [CrossRef]
- Zhang, Y.; Paul, E.M.; Sathyendra, V.; Davison, A.; Sharkey, N.; Bronson, S.; Srinivasan, S.; Gross, T.S.; Donahue, H.J. Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS ONE 2011, 6, e23516. [Google Scholar] [CrossRef] [Green Version]
- Watkins, M.; Grimston, S.K.; Norris, J.Y.; Guillotin, B.; Shaw, A.; Beniash, E.; Civitelli, R. Osteoblast connexin43 modulates skeletal architecture by regulating both arms of bone remodeling. Mol. Biol. Cell 2011, 22, 1240–1251. [Google Scholar] [CrossRef]
- Lin, F.X.; Zheng, G.Z.; Chang, B.; Chen, R.C.; Zhang, Q.H.; Xie, P.; Xie, D.; Yu, G.Y.; Hu, Q.X.; Liu, D.Z.; et al. Connexin 43 Modulates Osteogenic Differentiation of Bone Marrow Stromal Cells Through GSK-3beta/Beta-Catenin Signaling Pathways. Cell Physiol. Biochem. 2018, 47, 161–175. [Google Scholar] [CrossRef]
- Gupta, A.; Leser, J.M.; Gould, N.R.; Buo, A.M.; Moorer, M.C.; Stains, J.P. Connexin43 regulates osteoprotegerin expression via ERK1/2 -dependent recruitment of Sp1. Biochem. Biophys. Res. Commun. 2019, 509, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Nieto, D.; Li, L.; Kohler, A.; Ghiaur, G.; Ishikawa, E.; Sengupta, A.; Madhu, M.; Arnett, J.L.; Santho, R.A.; Dunn, S.K.; et al. Connexin-43 in the osteogenic BM niche regulates its cellular composition and the bidirectional traffic of hematopoietic stem cells and progenitors. Blood 2012, 119, 5144–5154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Q.; Frenette, P.S. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018, 48, 632–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [Green Version]
- Cordeiro Gomes, A.; Hara, T.; Lim, V.Y.; Herndler-Brandstetter, D.; Nevius, E.; Sugiyama, T.; Tani-Ichi, S.; Schlenner, S.; Richie, E.; Rodewald, H.R.; et al. Hematopoietic Stem Cell Niches Produce Lineage-Instructive Signals to Control Multipotent Progenitor Differentiation. Immunity 2016, 45, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Greenbaum, A.; Hsu, Y.M.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Winkler, I.G.; Barbier, V.; Nowlan, B.; Jacobsen, R.N.; Forristal, C.E.; Patton, J.T.; Magnani, J.L.; Levesque, J.P. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat. Med. 2012, 18, 1651–1657. [Google Scholar] [CrossRef]
- Zhang, F.; Cheng, J.; Lam, G.; Jin, D.K.; Vincent, L.; Hackett, N.R.; Wang, S.; Young, L.M.; Hempstead, B.; Crystal, R.G.; et al. Adenovirus vector E4 gene regulates connexin 40 and 43 expression in endothelial cells via PKA and PI3K signal pathways. Circ. Res. 2005, 96, 950–957. [Google Scholar] [CrossRef]
- Yuan, D.; Sun, G.; Zhang, R.; Luo, C.; Ge, M.; Luo, G.; Hei, Z. Connexin 43 expressed in endothelial cells modulates monocyteendothelial adhesion by regulating cell adhesion proteins. Mol. Med. Rep. 2015, 12, 7146–7152. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Day, K.H.; Damon, D.N.; Duling, B.R. Endothelial cell-specific knockout of connexin 43 causes hypotension and bradycardia in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 9989–9994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Zingler, M.; Harrison, J.K.; Scott, E.W.; Cogle, C.R.; Luo, D.; Raizada, M.K. Angiotensin II Regulation of Proliferation, Differentiation, and Engraftment of Hematopoietic Stem Cells. Hypertension 2016, 67, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Hooper, A.T.; Butler, J.M.; Nolan, D.J.; Kranz, A.; Iida, K.; Kobayashi, M.; Kopp, H.G.; Shido, K.; Petit, I.; Yanger, K.; et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell. 2009, 4, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Himburg, H.A.; Termini, C.M.; Schlussel, L.; Kan, J.; Li, M.; Zhao, L.; Fang, T.; Sasine, J.P.; Chang, V.Y.; Chute, J.P. Distinct Bone Marrow Sources of Pleiotrophin Control Hematopoietic Stem Cell Maintenance and Regeneration. Cell Stem Cell. 2018, 23, 370–381.e5. [Google Scholar] [CrossRef] [Green Version]
- Chute, J.P.; Muramoto, G.G.; Salter, A.B.; Meadows, S.K.; Rickman, D.W.; Chen, B.; Himburg, H.A.; Chao, N.J. Transplantation of vascular endothelial cells mediates the hematopoietic recovery and survival of lethally irradiated mice. Blood 2007, 109, 2365–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramoto, G.G.; Chen, B.; Cui, X.; Chao, N.J.; Chute, J.P. Vascular endothelial cells produce soluble factors that mediate the recovery of human hematopoietic stem cells after radiation injury. Biol. Blood Marrow. Transplant. 2006, 12, 530–540. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Poulos, M.G.; Palikuqi, B.; Badwe, C.R.; Lis, R.; Kunar, B.; Ding, B.S.; Rabbany, S.Y.; Shido, K.; Butler, J.M.; et al. Endothelial jagged-2 sustains hematopoietic stem and progenitor reconstitution after myelosuppression. J. Clin. Invest. 2017, 127, 4242–4256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Liu, Y.; Jeong, H.W.; Stehling, M.; Dinh, V.V.; Zhou, B.; Adams, R.H. Apelin(+) Endothelial Niche Cells Control Hematopoiesis and Mediate Vascular Regeneration after Myeloablative Injury. Cell Stem Cell. 2019, 25, 768–783.e6. [Google Scholar] [CrossRef] [Green Version]
- Itkin, T.; Gur-Cohen, S.; Spencer, J.A.; Schajnovitz, A.; Ramasamy, S.K.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; Poulos, M.G.; et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef]
- Winkler, I.G.; Sims, N.A.; Pettit, A.R.; Barbier, V.; Nowlan, B.; Helwani, F.; Poulton, I.J.; van Rooijen, N.; Alexander, K.A.; Raggatt, L.J.; et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 2010, 116, 4815–4828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, A.; Lucas, D.; Hidalgo, A.; Mendez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; van Rooijen, N.; et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Pettit, A.R.; Chang, M.K.; Hume, D.A.; Raggatt, L.J. Osteal macrophages: A new twist on coupling during bone dynamics. Bone 2008, 43, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A.; Branes, M.C.; Berman, J.W.; Saez, J.C. TNF-alpha plus IFN-gamma induce connexin43 expression and formation of gap junctions between human monocytes/macrophages that enhance physiological responses. J. Immunol. 2003, 170, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Anand, R.J.; Dai, S.; Rippel, C.; Leaphart, C.; Qureshi, F.; Gribar, S.C.; Kohler, J.W.; Li, J.; Stolz, D.B.; Sodhi, C.; et al. Activated macrophages inhibit enterocyte gap junctions via the release of nitric oxide. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G109–G119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matemba, S.F.; Lie, A.; Ransjo, M. Regulation of osteoclastogenesis by gap junction communication. J. Cell Biochem. 2006, 99, 528–537. [Google Scholar] [CrossRef]
- Kylmaoja, E.; Nakamura, M.; Kokkonen-Puupera, H.; Ronkainen, V.P.; Lehenkari, P.; Tuukkanen, J. Gap junctional communication is involved in differentiation of osteoclasts from bone marrow and peripheral blood monocytes. Heliyon 2018, 4, e00621. [Google Scholar] [CrossRef] [Green Version]
- Pacheco-Costa, R.; Hassan, I.; Reginato, R.D.; Davis, H.M.; Bruzzaniti, A.; Allen, M.R.; Plotkin, L.I. High bone mass in mice lacking Cx37 because of defective osteoclast differentiation. J. Biol. Chem. 2014, 289, 8508–8520. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.W.; Christen, T.; Roth, I.; Chadjichristos, C.E.; Derouette, J.P.; Foglia, B.F.; Chanson, M.; Goodenough, D.A.; Kwak, B.R. Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 2006, 12, 950–954. [Google Scholar] [CrossRef]
- Chang, K.H.; Sengupta, A.; Nayak, R.C.; Duran, A.; Lee, S.J.; Pratt, R.G.; Wellendorf, A.M.; Hill, S.E.; Watkins, M.; Gonzalez-Nieto, D.; et al. p62 is required for stem cell/progenitor retention through inhibition of IKK/NF-kappaB/Ccl4 signaling at the bone marrow macrophage-osteoblast niche. Cell Rep. 2014, 9, 2084–2097. [Google Scholar] [CrossRef] [Green Version]
- Albiero, M.; Poncina, N.; Ciciliot, S.; Cappellari, R.; Menegazzo, L.; Ferraro, F.; Bolego, C.; Cignarella, A.; Avogaro, A.; Fadini, G.P. Bone Marrow Macrophages Contribute to Diabetic Stem Cell Mobilopathy by Producing Oncostatin M. Diabetes 2015, 64, 2957–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, J.J.; Marlein, C.R.; Moore, J.A.; Hellmich, C.; Wojtowicz, E.E.; Smith, J.G.W.; Macaulay, I.; Sun, Y.; Morfakis, A.; Patterson, A.; et al. ROS-mediated PI3K activation drives mitochondrial transfer from stromal cells to hematopoietic stem cells in response to infection. Proc. Natl. Acad. Sci. USA 2019, 116, 24610–24619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Golan, K.; Althoff, M.; Petrovich-Kopitman, E.; Wellendorf, A.; Mohmoud, F.; Bertagna, M.; Gonzalez-Nieto, D.; Barrio, L.C.; Lapidot, T.; et al. Bone Marrow Hematopoietic Connexin 43 Is Required for Mitotransfer and AMPK Dependent Mesenchymal Microenvironment Regeneration after Irradiation. Blood 2018, 132, 872. [Google Scholar] [CrossRef]
- Lemoli, R.M.; Ferrari, D.; Fogli, M.; Rossi, L.; Pizzirani, C.; Forchap, S.; Chiozzi, P.; Vaselli, D.; Bertolini, F.; Foutz, T.; et al. Extracellular nucleotides are potent stimulators of human hematopoietic stem cells in vitro and in vivo. Blood 2004, 104, 1662–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, L.; Salvestrini, V.; Ferrari, D.; Di Virgilio, F.; Lemoli, R.M. The sixth sense: Hematopoietic stem cells detect danger through purinergic signaling. Blood 2012, 120, 2365–2375. [Google Scholar] [CrossRef] [Green Version]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Paraguassu-Braga, F.H.; Borojevic, R.; Bouzas, L.F.; Barcinski, M.A.; Bonomo, A. Bone marrow stroma inhibits proliferation and apoptosis in leukemic cells through gap junction-mediated cell communication. Cell Death Differ. 2003, 10, 1101–1108. [Google Scholar] [CrossRef]
- Desbourdes, L.; Javary, J.; Charbonnier, T.; Ishac, N.; Bourgeais, J.; Iltis, A.; Chomel, J.C.; Turhan, A.; Guilloton, F.; Tarte, K.; et al. Alteration Analysis of Bone Marrow Mesenchymal Stromal Cells from De Novo Acute Myeloid Leukemia Patients at Diagnosis. Stem Cells Dev. 2017, 26, 709–722. [Google Scholar] [CrossRef]
- Bendall, L.J.; Daniel, A.; Kortlepel, K.; Gottlieb, D.J. Bone marrow adherent layers inhibit apoptosis of acute myeloid leukemia cells. Exp. Hematol. 1994, 22, 1252–1260. [Google Scholar]
- Van Gosliga, D.; Schepers, H.; Rizo, A.; van der Kolk, D.; Vellenga, E.; Schuringa, J.J. Establishing long-term cultures with self-renewing acute myeloid leukemia stem/progenitor cells. Exp. Hematol. 2007, 35, 1538–1549. [Google Scholar] [CrossRef]
- Ailles, L.E.; Gerhard, B.; Hogge, D.E. Detection and characterization of primitive malignant and normal progenitors in patients with acute myelogenous leukemia using long-term coculture with supportive feeder layers and cytokines. Blood 1997, 90, 2555–2564. [Google Scholar] [CrossRef]
- Griessinger, E.; Anjos-Afonso, F.; Pizzitola, I.; Rouault-Pierre, K.; Vargaftig, J.; Taussig, D.; Gribben, J.; Lassailly, F.; Bonnet, D. A niche-like culture system allowing the maintenance of primary human acute myeloid leukemia-initiating cells: A new tool to decipher their chemoresistance and self-renewal mechanisms. Stem Cells Transl. Med. 2014, 3, 520–529. [Google Scholar] [CrossRef]
- Kouzi, F.; Zibara, K.; Bourgeais, J.; Picou, F.; Gallay, N.; Brossaud, J.; Dakik, H.; Roux, B.; Hamard, S.; Le Nail, L.R.; et al. Disruption of gap junctions attenuates acute myeloid leukemia chemoresistance induced by bone marrow mesenchymal stromal cells. Oncogene 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connors, B.W. Tales of a dirty drug: Carbenoxolone, gap junctions, and seizures. Epilepsy Curr. 2012, 12, 66–68. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.C.; Tykocinski, M.L. Bone marrow stromal cell blockade of human leukemic cell differentiation. Blood 1994, 83, 2221–2229. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Chen, Y.; Wen, L.; Yang, L.; Cui, G. Expression of connexin 32 and connexin 43 in acute myeloid leukemia and their roles in proliferation. Oncol. Lett. 2012, 4, 1003–1007. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wen, Q.; Chen, X.L.; Yang, S.J.; Gao, L.; Gao, L.; Zhang, C.; Li, J.L.; Xiang, X.X.; Wan, K.; et al. All-trans retinoic acid arrests cell cycle in leukemic bone marrow stromal cells by increasing intercellular communication through connexin 43-mediated gap junction. J. Hematol. Oncol. 2015, 8, 110. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, X.; Li, Z.J.; Chen, X.H. Up-regulation of Cx43 expression and GJIC function in acute leukemia bone marrow stromal cells post-chemotherapy. Leuk. Res. 2010, 34, 631–640. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Y.; Si, Y.J.; Chen, X.H.; Li, Z.J.; Gao, L.; Gao, L.; Zhang, C. Effect of Cx43 gene-modified leukemic bone marrow stromal cells on the regulation of Jurkat cell line in vitro. Leuk. Res. 2012, 36, 198–204. [Google Scholar] [CrossRef]
- Yang, S.; Wen, Q.; Liu, Y.; Zhang, C.; Wang, M.; Chen, G.; Gong, Y.; Zhong, J.; Chen, X.; Stucky, A.; et al. Increased expression of CX43 on stromal cells promotes leukemia apoptosis. Oncotarget 2015, 6, 44323–44331. [Google Scholar] [CrossRef] [Green Version]
- Sinyuk, M.; Alvarado, A.G.; Nesmiyanov, P.; Shaw, J.; Mulkearns-Hubert, E.E.; Eurich, J.T.; Hale, J.S.; Bogdanova, A.; Hitomi, M.; Maciejewski, J.; et al. Cx25 contributes to leukemia cell communication and chemosensitivity. Oncotarget 2015, 6, 31508–31521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Xu, Y.B.; Wang, Q.; Lu, Y.; Zheng, Y.; Wang, Y.C.; Lubbert, M.; Zhao, K.W.; Chen, G.Q. Leukemogenic AML1-ETO fusion protein upregulates expression of connexin 43: The role in AML 1-ETO-induced growth arrest in leukemic cells. J. Cell. Physiol. 2006, 208, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Ryningen, A.; Saeterdal, L.R.; Nepstad, I.; Foss, B.; Bruserud, O. Connexin expression in human acute myeloid leukemia cells: Identification of patient subsets based on protein and global gene expression profiles. Int. J. Mol. Med. 2015, 35, 645–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Sun, Y.; Wang, Z.; Huang, Z.; Li, B.; Fu, J. Up-regulation of connexin-43 expression in bone marrow mesenchymal stem cells plays a crucial role in adhesion and migration of multiple myeloma cells. Leuk Lymphoma 2015, 56, 211–218. [Google Scholar] [CrossRef]
- Fu, J. Cx43 expressed on bone marrow stromal cells plays an essential role in multiple myeloma cell survival and drug resistance. Arch. Med. Sci. 2017, 13, 236–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 2017, 6, e2187. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Oeding, S.J.; Majstrowicz, K.; Hu, X.P.; Schwarz, V.; Freitag, A.; Honnert, U.; Nikolaus, P.; Bahler, M. Identification of Miro1 and Miro2 as mitochondrial receptors for myosin XIX. J. Cell Sci. 2018, 131, jcs219469. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Fan, X.L.; Jiang, D.; Zhang, Y.; Li, X.; Xu, Z.B.; Fang, S.B.; Chiu, S.; Tse, H.F.; Lian, Q.; et al. Connexin 43-Mediated Mitochondrial Transfer of iPSC-MSCs Alleviates Asthma Inflammation. Stem Cell Rep. 2018, 11, 1120–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, J.T.; Parker, I.; Smith, I.F. Communication of Ca2+ signals via tunneling membrane nanotubes is mediated by transmission of inositol trisphosphate through gap junctions. Cell Calcium 2016, 60, 266–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahrouf-Yorgov, M.; Augeul, L.; Da Silva, C.C.; Jourdan, M.; Rigolet, M.; Manin, S.; Ferrera, R.; Ovize, M.; Henry, A.; Guguin, A.; et al. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ. 2017, 24, 1224–1238. [Google Scholar] [CrossRef] [Green Version]
- Boultwood, J.; Fidler, C.; Mills, K.I.; Frodsham, P.M.; Kusec, R.; Gaiger, A.; Gale, R.E.; Linch, D.C.; Littlewood, T.J.; Moss, P.A.; et al. Amplification of mitochondrial DNA in acute myeloid leukaemia. Br. J. Haematol. 1996, 95, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [Green Version]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [Green Version]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Raso-Barnett, L.; Scott, M.A.; Ingham, C.J.; Collins, A.; Bowles, K.M.; Rushworth, S.A. PGC-1alpha driven mitochondrial biogenesis in stromal cells underpins mitochondrial trafficking to leukemic blasts. Leukemia 2018, 32, 2073–2077. [Google Scholar] [CrossRef]
- Polak, R.; de Rooij, B.; Pieters, R.; den Boer, M.L. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef] [Green Version]
- Forte, D.; García-Fernández, M.; Sánchez-Aguilera, A.; Stavropoulou, V.; Fielding, C.; Martín-Pérez, D.; Tzankov, A.; Schwaller, J.; Méndez-Ferrer, S. Leukemic Stem Cells Co-Opt Normal Bone Marrow Niches as a Source of Energy and Antioxidant Defence. Blood 2017, 130, 94. [Google Scholar]
- Bodi, E.; Hurtado, S.P.; Carvalho, M.A.; Borojevic, R.; Carvalho, A.C. Gap junctions in hematopoietic stroma control proliferation and differentiation of blood cell precursors. An. Acad. Bras. Cienc. 2004, 76, 743–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veliz, L.P.; Gonzalez, F.G.; Duling, B.R.; Saez, J.C.; Boric, M.P. Functional role of gap junctions in cytokine-induced leukocyte adhesion to endothelium in vivo. Am. J. Physiol. Heart. Circ. Physiol. 2008, 295, H1056–H1066. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cell Type | Connexin Type | Function | Ref. |
|---|---|---|---|
| Normal HSC and BM Stromal Cells | |||
| LT-HSC and progenitors | Cx43, Cx45, Cx31, Cx31.1, Cx32, Cx37, and Cx50 | Expression | [15,28] |
| LSK and LK cells | Cx32 | Maintain HSPC quiescence and stemness | [29] |
| HSPC | Cx43 | Reduces HSC senescence via ROS transfer to BMSC during stress induced hematopoietic regeneration | [14,15] |
| BM stromal cells (Osteoblasts, MSC and endothelial cells) | Cx31 | Expression | [93] |
| BM stromal cells (Osteoblasts, MSC and endothelial cells) | Cx43 and Cx45 | Regulates CXCL12 secretion, HSC growth, differentiation and homing | [8,17,112,142] |
| BM stromal cells | Cx43 | 1) Determine bone mass and bone mineral density by modulating osteogenesis | [16,110,142,182] |
| 2) Mitochondria transfer from BMSC to HSC and emergency granulopoiesis | |||
| 3) HSPC proliferation and differentiation of myeloid blood cell precursor | |||
| 4) Hematopoietic regeneration after chemotherapy | |||
| BM Endothelial cells | Cx43 | Normal vascular function, leukocyte adhesion and transmigration | [9,183] |
| Leukemia Cell Lines, Primary Cells, and Leukemic BM Stromal Cells | |||
| OCIM2 and OCI-AML3 cells | Cx43 and Cx32 | ↑Cell proliferation | [156] |
| CCRF-CEM lymphoblast cells | Cx33, Cx40, Cx43, Cx45, Cx46, and Cx50 | ↑ Chemoresistance | [147] |
| ↓Apoptosis | |||
| ↓Differentiation | |||
| HL-60 and PBL-985 cells | GJ | ↓Differentiation | [155] |
| U937 AML cells expressing AML1-ETO fusion protein | Cx43 | ↓Cell proliferation | [162] |
| KG-1, KG-1a, HL-60, OCI-AML3, MV4-11, MoLM-13 Jurkat, and THP1 cells | Cx25, Cx31.9, Cx40, Cx43, Cx45, and Cx59 | ↑Cell proliferation | [161] |
| ↑Chemoresistance | |||
| Primary AML cells | Cx26, Cx32, Cx37, Cx43, and Cx45 | ↑ Chemoresistance | [163] |
| ↓Apoptosis | |||
| AML-blasts and BM CD34+ cells | Cx43, Cx45, Cx25, Cx31.9, and Cx59 | ↑Cell proliferation | [153,163] |
| ↑Chemoresistance | |||
| AML BM-stromal cells | Cx25, Cx26, Cx30, Cx31, Cx32, Cx36, Cx37, Cx40, Cx46, and Cx62 | ↑Cell proliferation | [153] |
| ↑Chemoresistance | |||
| Primary Multiple Myeloma (MM) cells and cell lines (RPMI 8226, U266, and XG-7) | Cx43 | Adhesion and migration of MM cells | [164,165] |
| ↑Cell proliferation | |||
| ↑Chemoresistance | |||
| Multiple Myeloma BM-stromal cells | Cx43 | Adhesion and migration of MM cells | [164,165] |
| ↑Cell proliferation | |||
| ↑Chemoresistance | |||
| Cx32-KO mice | ↑Leukemia incidence | [76] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, A.K.; Cancelas, J.A. Gap Junctions in the Bone Marrow Lympho-Hematopoietic Stem Cell Niche, Leukemia Progression, and Chemoresistance. Int. J. Mol. Sci. 2020, 21, 796. https://doi.org/10.3390/ijms21030796
Singh AK, Cancelas JA. Gap Junctions in the Bone Marrow Lympho-Hematopoietic Stem Cell Niche, Leukemia Progression, and Chemoresistance. International Journal of Molecular Sciences. 2020; 21(3):796. https://doi.org/10.3390/ijms21030796
Chicago/Turabian StyleSingh, Abhishek K., and Jose A. Cancelas. 2020. "Gap Junctions in the Bone Marrow Lympho-Hematopoietic Stem Cell Niche, Leukemia Progression, and Chemoresistance" International Journal of Molecular Sciences 21, no. 3: 796. https://doi.org/10.3390/ijms21030796
APA StyleSingh, A. K., & Cancelas, J. A. (2020). Gap Junctions in the Bone Marrow Lympho-Hematopoietic Stem Cell Niche, Leukemia Progression, and Chemoresistance. International Journal of Molecular Sciences, 21(3), 796. https://doi.org/10.3390/ijms21030796