A History of Molecular Chaperone Structures in the Protein Data Bank

Abstract
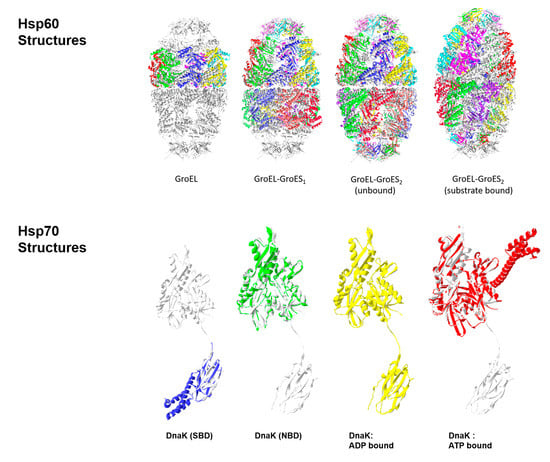
:
1. Introduction
2. Review
2.1. Hsp60s (Chaperonins/Heptameric Anfinsen Cages)
2.2. Hsp70s (Multidomain Chaperone Systems)
2.3. Hsp90s (Receptor Protein Refinement)
2.4. Hsp104s (Disaggregating Complexes)
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Epstein, C.J.; Goldberger, R.F.; Anfinsen, C.B. Genetic control of tertiary protein structure—studies with model systems. Cold Spring Harb. Symp. 1963, 28, 439–449. [Google Scholar] [CrossRef]
- Finka, A.; Mattoo, R.U.H.; Goloubinoff, P. Experimental Milestones in the Discovery of Molecular Chaperones as Polypeptide Unfolding Enzymes. Annu. Rev. Biochem. 2016, 85, 715–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finka, A.; Goloubinoff, P. Proteomic data from human cell cultures refine mechanisms of chaperone mediated protein homeostasis. Cell Stress Chaperones. 2013, 18, 591–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternberg, N. Properties of a mutant Escherichia coli defective in bacteriophage λ head formation (groE): II. The propagation of phage λ. J. Mol. Biol. 1973, 76, 25–44. [Google Scholar] [CrossRef]
- Goloubinoff, P.; Christeller, J.T.; Gatenby, A.A.; Lorimer, G.H. Reconstitution of active dimeric ribulose bisphosphate carboxylase from an unfolded state depends on two chaperonin proteins and Mg-ATP. Nature. 1989, 342, 884–889. [Google Scholar] [CrossRef]
- Skowyra, D.; Georgopoulos, C.; Zylicz, M. The E. coli dnaK gene product, the hsp70 homolog, can reactivate heat-inactivated RNA polymerase in an ATP hydrolysis-dependent manner. Cell 1990, 62, 939–944. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Hohn, T.; Hohn, B.; Engel, A.; Wurtz, M.; Smith, P.R. Isolation and characterization of the host protein groE involved in bacteriophage lambda assembly. J. Mol. Biol. 1979, 129, 359–373. [Google Scholar] [CrossRef]
- Hemmingsen, S.M.; Woolford, C.; van der Vies, S.M.; Tilly, K.; Dennis, D.T.; Georgopoulos, C.P.; Hendrix, R.W.; Ellis, R.J. Homologous plant and bacterial proteins chaperone oligomeric protein assembly. Nature 1998, 26, 330–334. [Google Scholar] [CrossRef]
- Viitanen, P.V.; Lubben, T.H.; Reed, J.; Goloubinoff, P.; O’Keefe, D.P.; Lorimer, G.H. Chaperonin-facilitated refolding of ribulosebisphosphate car- boxylase and ATP hydrolysis by chaperonin 60 (groEL) are K + dependent. Biochemistry 1990, 29, 5665–5671. [Google Scholar] [CrossRef] [PubMed]
- Braig, K.; Otwinowski, Z.; Hegde, R.; Boisvert, D.C.; Joachimiak, A.; Horwich, A.L.; Sigler, P.B. The crystal structure of the bacterial chaperonin GroEL at 2.8 A. Nature. 1994, 371, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Horwich, A.L.; Sigler, P.B. The crystal structure of the asymmetric GroEL-GroES-(ADP) 7 chaperonin complex. Nature 1997, 388, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Landry, S.J.; Zeilstra-Ryalls, J.; Fayet, O.; Georgopoulos, C.; Gierasch, L.M. Characterization of a Functionally Important Mobile Domain of GroES. Nature. 1993, 364, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Nisemblat, S.; Yaniv, O.; Parnas, A.; Frolow, F.; Azem, A. Crystal structure of the human mitochondrial chaperonin symmetrical football complex. Proc. Natl. Acad. Sci. USA 2015, 112, 6044–6049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, X.; Ye, X.; La Ronde, N.A.; Lorimer, G.H. Crystal structure of the Football-shaped GroEL-GroES2-(ADPBeFx) 14 complex. Proc. Natl. Acad. Sci. USA 2014, 111, 12775–12780. [Google Scholar] [CrossRef] [Green Version]
- Koike-Takeshita, A.; Arakawa, T.; Taguchi, H.; Shimamura, T. Crystal Structure of a Symmetric Football-Shaped GroEL:GroES(2)-ATP(14) Complex Determined at 3.8 angstrom Reveals Rearrangement between Two GroEL Rings. J. Mol. Biol. 2014, 426, 3634–3641. [Google Scholar] [CrossRef] [Green Version]
- Buckle, A.M.; Zahn, R.; Fersht, A.R. A structural model for GroEL-polypeptide recognition. Proc. Natl. Acad. Sci. USA 1997, 94, 3571–3575. [Google Scholar] [CrossRef] [Green Version]
- Walsh, M.A.; Dementieva, I.; Evans, G.; Sanishvili, R.; Joachimiak, A. Taking MAD to the extreme: Ultrafast protein structure determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999, 55, 1168–1173. [Google Scholar] [CrossRef]
- Swain, J.F.; Dinler, G.; Sivendran, R.; Montgomery, D.L.; Stotz, M.; Gierasch, L.M. Hsp70 Chaperone Ligands Control Domain Association via an Allosteric Mechanism Mediated by the Interdomain Linker. Mol. Cell 2007, 26, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. CMLS 2005, 62, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Zhao, X.; Burkholder, W.F.; Gragerov, A.; Ogata, C.M.; Gottesman, M.E.; Hendrickson, W.A. Structural analysis of substrate binding by the molecular chaperone DnaK. Science 1996, 272, 1606–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Kurochkin, A.V.; Pang, Y.; Hu, W.; Flynn, G.C.; Zuiderweg, E.R. NMR solution structure of the 21 kDa chaperone protein DnaK substrate binding domain: A preview of chaperone-protein interaction. Biochemistry 1998, 37, 7929–7940. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, M.; Montgomery, D.L.; Stevens, S.Y.; Vander Kooi, C.W.; Feng, H.P.; Gierasch, L.M.; Zuiderweg, E.R. Structural insights into substrate binding by the molecular chaperone DnaK. Nat. Struct. Mol. Biol. 2000, 7, 298–303. [Google Scholar] [CrossRef]
- Stevens, S.Y.; Cai, S.; Pellecchia, M.; Zuiderweg, E.R. The solution structure of the bacterial HSP70 chaperone protein domain DnaK (393–507) in complex with the peptide NRLLLTG. Protein Sci. 2003, 12, 2588–2596. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J.; Hayer-Hartl, M.; Di Liberto, M.; Hartl, F.; Kuriyan, J. Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science 1997, 276, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-W.; Sun, Y.-J.; Wang, C.; Hsiao, C.-D. Crystal Structures of the 70-kDa Heat Shock Proteins in Domain Disjoining Conformation. J. Biol. Chem. 2008, 283, 15502. [Google Scholar] [CrossRef] [Green Version]
- Liebscher, M.; Roujeinikova, A. Allosteric coupling between the lid and interdomain linker in DnaK revealed by inhibitor binding studies. J. Bacteriol. 2009, 191, 1456–1462. [Google Scholar] [CrossRef] [Green Version]
- Bertelsen, E.B.; Chang, L.; Gestwicki, J.E.; Zuiderweg, E.R. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA 2009, 106, 8471–8476. [Google Scholar] [CrossRef] [Green Version]
- Kityk, R.; Kopp, J.; Mayer, M.P. Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones. Mol. Cell 2018, 69, 227–237.e4. [Google Scholar] [CrossRef]
- Qi, R.; Sarbeng, E.B.; Liu, Q.; Le, K.Q.; Xu, X.; Xu, H.; Liu, Q. Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat. Struct. Mol. Biol. 2013, 20, 900–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-C.; Naveen, V.; Chien, C.-H.; Chang, Y.-W.; Hsiao, C.-D. Crystal Structure of Dnak Protein Complexed with Nucleotide Exchange Factor Grpe in Dnak Chaperone System: Insight into Intermolecular Communication. J. Biol. Chem. 2012, 287, 21461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Flanagan, J.M.; Prestegard, J.H. The influence of C-terminal extension on the structure of the “J-domain” in E. coli DnaJ. Protein Sci. 1999, 8, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Qian, X.; Sha, B. The crystal structure of the yeast Hsp40 Ydj1 complexed with its peptide substrate. Structure 2003, 11, 1475–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horne, B.E.; Li, T.; Genevaux, P.; Georgopoulos, C.; Landry, S.J. The Hsp40 J-domain stimulates Hsp70 when tethered by the client to the ATPase domain. J. Biol Chem. 2010, 285, 21679–21688. [Google Scholar] [CrossRef] [Green Version]
- Greene, M.K.; Maskos, K.; Landry, S.J. Role of the J-domain in the cooperation of Hsp40 with Hsp70. Proc. Natl. Acad. Sci. USA 1998, 95, 6108–6113. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Buchner, J. Structure, function and regulation of the hsp90 machinery. Biomed. J. 2013, 36, 106–117. [Google Scholar] [CrossRef]
- Prodromou, C.; Roe, S.M.; Piper, P.W.; Pearl, L.H. A molecular clamp in the crystal structure of the N-terminal domain of the yeast Hsp90 chaperone. Nat. Struct. Mol. Biol. 1997, 4, 477–482. [Google Scholar] [CrossRef]
- Meyer, P.; Prodromou, C.; Hu, B.; Vaughan, C.; Roe, S.M.; Panaretou, B.; Pearl, L.H. Structural and Functional Analysis of the Middle Segment of Hsp90. Implications for ATP Hydrolysis and Client Protein and Cochaperone Interactions. Mol. Cell 2003, 11, 647. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.F.; Shiau, A.K.; Agard, D.A. The crystal structure of the carboxy-terminal dimerization domain of htpG, the Escherichia coli Hsp90, reveals a potential substrate binding site. Structure 2004, 12, 1087–1097. [Google Scholar] [CrossRef] [Green Version]
- Huai, Q.; Wang, H.; Liu, Y.; Kim, H.Y.; Toft, D.; Ke, H. Structures of the N-terminal and middle domains of E. coli Hsp90 and conformation changes upon ADP binding. Structure 2005, 13, 579–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiau, A.K.; Harris, S.F.; Southworth, D.R.; Agard, D.A. Structural Analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell 2006, 127, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.; Meyer, P.; Panaretou, B.; Piper, P.W.; Pearl, L.H. Crystal Structure of an Hsp90-Nucleotide-P23/Sba1 Closed Chaperone Complex. Nature 2006, 440, 1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, S.M.; Ali, M.M.U.; Meyer, P.; Vaughan, C.K.; Panaretou, B.; Piper, P.W.; Pearl, L.H. The Mechanism of Hsp90 Regulation by the Protein Kinase-Specific Cochaperone p50 (Cdc37). Cell 2004, 116, 87. [Google Scholar] [CrossRef] [Green Version]
- Verba, K.A.; Wang, R.Y.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic Structure of Hsp90-Cdc37-Cdk4 Reveals that Hsp90 Traps and Stabilizes an Unfolded Kinase. Science 2016, 352, 1542. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Casais, C.; Ichimura, K.; Shirasu, K. HSP90 interacts with RAR1 and SGT1 and is essential for RPS2-mediated disease resistance in Arabidopsis. Proc. Natl. Acad. Sci. USA 2003, 100, 11777–11782. [Google Scholar] [CrossRef] [Green Version]
- Kakihara, Y.; Houry, W.A. The R2TP complex: Discovery and functions. Biochimica et Biophysica Acta 2012, 1823, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Boulon, S.; Pradet-Balade, B.; Verheggen, C.; Molle, D.; Boireau, S.; Georgieva, M.; Bertrand, E. HSP90 and its R2TP/Prefoldin-like cochaperone are involved in the cytoplasmic assembly of RNA polymerase II. Mol. Cell 2010, 39, 912–924. [Google Scholar] [CrossRef] [Green Version]
- Kandror, O.; Bretschneider, N.; Kreydin, E.; Cavalieri, D.; Goldberg, A.L. Yeast adapt to near-freezing temperatures by STRE/Msn2, 4-dependent induction of trehalose synthesis and certain molecular chaperones. Mol. Cell 2004, 13, 771–781. [Google Scholar] [CrossRef]
- Parsell, D.A.; Lindquist, S. The function of heat-shock proteins in stress tolerance: Degradation and reactivation of damaged proteins. Annu. Rev. Genet. 1993, 27, 437–496. [Google Scholar] [CrossRef]
- Godon, C.; Lagniel, G.; Lee, J.; Buhler, J.M.; Kieffer, S.; Perrot, M.; Labarre, J. The H2O2 stimulon in Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 22480–22489. [Google Scholar] [CrossRef] [Green Version]
- Borchsenius, A.S.; Muller, S.; Newnam, G.P.; Inge-Vechtomov, S.G.; Chernoff, Y.O. Prion variant maintained only at high levels of the Hsp104 disaggregase. Curr. Genet. 2006, 49, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sha, B. Crystal structure of E. coli Hsp100 ClpB nucleotide-binding domain 1 (NBD1) and mechanistic studies on ClpB ATPase activity. J. Mol. Biol. 2002, 318, 1127–1137. [Google Scholar] [CrossRef]
- Lee, S.; Sowa, M.E.; Watanabe, Y.; Sigler, P.B.; Chiu, W.; Yoshida, M.; Tsai, F.T.F. The Structure of ClpB: A Molecular Chaperone that Rescues Proteins from an Aggregated State. Cell 2003, 115, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Biter, A.B.; Lee, S.; Sung, N.; Tsai, F.T. Structural basis for intersubunit signaling in a protein disaggregating machine. Proc. Natl. Acad. Sci. USA 2012, 109, 12515–12520. [Google Scholar] [CrossRef] [Green Version]
- Zeymer, C.; Barends, T.R.M.; Werbeck, N.D.; Schlichting, I.; Reinstein, J. Elements in nucleotide sensing and hydrolysis of the AAA+ disaggregation machine ClpB: A structure-based mechanistic dissection of a molecular motor. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 582–595. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Li, J.; Weaver, C.; Lucius, A.; Sha, B. Crystal structures of Hsp104 N-terminal domains from Saccharomyces cerevisiae and Candida albicans suggest the mechanism for the function of Hsp104 in dissolving prions. Acta Crystallogr. D Struct. Biol. 2017, 73, 365–372. [Google Scholar] [CrossRef]
- Gates, S.N.; Yokom, A.L.; Lin, J.; Jackrel, M.E.; Rizo, A.N.; Kendsersky, N.M.; Southworth, D.R. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 2017, 357, 273–279. [Google Scholar] [CrossRef] [Green Version]
- Deville, C.; Carroni, M.; Franke, K.B.; Topf, M.; Bukau, B.; Mogk, A.; Saibil, H.R. Structural pathway of regulated substrate transfer and threading through an Hsp100 disaggregase. Sci. Adv. 2017, 3, e1701726. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.D.; Schumacher, R.J.; Ross, E.D.; Toft, D.O. Hop modulates Hsp70/Hsp90 interactions in protein folding. J. Biol. Chem. 1998, 273, 3679–3686. [Google Scholar] [CrossRef] [Green Version]
- Kyratsous, C.A.; Silverstein, S.J.; DeLong, C.R.; Panagiotidis, C.A. Chaperone-fusion expression plasmid vectors for improved solubility of recombinant proteins in Escherichia coli. Gene 2009, 440, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chaperone Family | Functions | Annotated Subcellular Localizations (UniProt [8]) * | Curated Samples and Related Proteins: UniProt [8] *,++ |
|---|---|---|---|
| Hsp60 | Segregate unfolded polypeptide chains Promote unfolding of misfolded polypeptides by both active and passive mechanisms | Chloroplast Cytoplasm Mitochondria | Prokaryotic: 60 kDa chaperonin (1–4); cpn60; groEL; groL; groL1 to groL5; mimG; prmG; thsC Eukaryotic: cpn60 (I and II; 1 and 2); groL(A and B); hsp60; Hspd1; Rubisco large subunit binding protein (alpha1, beta1, and beta2) Tcm62 |
| Hsp70 | Unfold misfolded polypeptides Translocate unfolded polyproteins through membranes Dissociate protein complexes | Chloroplast Cytoplasm Endoplasmic Reticulum (ER) Mitochondria Nucleus | Viral: Movement protein Hsp70h Prokaryotic: Heat shock 70 kDA protein; Hsp70; chaperone protein DnaK (1–3): [Cochaperones: DnaJ; GrpE (1 and 2)] HscA: [Cochaperone: HscB] HscC; Ssa (1 and 2); SsC1 Eukaryotic: Heat shock 70 kDA protein (1, 1A, 1B, 2, 3, 4, 4L, 5–10, 12–18) Chaperone Protein DnaK [Cochaperone: DnaJ] AtHsp70- (2,4,12,13); endoplasmic reticulum chaperone: BiP (1–5, 8); heat shock 70 kDA protein cognate (1, 2, 4, 5, II, IV); Hsc70; heat shock protein 70.2; Hsp1; hypoxia upregulated protein 1 (hyou1); Lhs1 major heat shock 70 kDA protein (Aa, Ab, Ba, Bb, Bbb, Bb); Sse (1 and 2); SsA (1–3); SsB1; SsC (1, 3); SsQ1; Sce70 |
| Hsp90 | Modification of kinases, steroid hormone receptors, and transcription factors | Cytoplasm Endoplasmic Reticulum (ER) Mitochondria Nucleus | Prokaryotic: Chaperone protein HtpG Eukaryotic: ATP-dependent molecular chaperone Hsc82; endoplasmin; endoplasmin homologs (Lpg3, Hsp90-7, grp94); heat shock cognate protein 80; heat shock-like 85 kDa protein; heat shock protein 81 (1–3); Hsp83; Hsp90 (1-6, alpha, A2, beta): [Cochaperones: Aha1; AHSA2P (putative); Cdc37; Cdc37-like 1; daf-41; Hch1; Hsp interacting protein, HIP; p23 (1, 2); PhPL3; peptidyl-prolyl cis trans isomerases (PPID, FKBP (4, 5, 8, 62, 65)); Protein canopy homolog 3; protein disulfide isomerase (PDI-A2, A4, A6); PPP5C; Sba1; Sgt (a, 1); OsSGT1, AtSgt1; Shu; Unc45-A,B (potential); wos2; Mod-E; Swo1 |
| Hsp104 | Dissociation, refolding, and resolubilization of protein aggregates [8] | Cytoplasm Nucleus | Eukaryotic: ClpB1 Hsp104 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bascos, N.A.D.; Landry, S.J. A History of Molecular Chaperone Structures in the Protein Data Bank. Int. J. Mol. Sci. 2019, 20, 6195. https://doi.org/10.3390/ijms20246195
Bascos NAD, Landry SJ. A History of Molecular Chaperone Structures in the Protein Data Bank. International Journal of Molecular Sciences. 2019; 20(24):6195. https://doi.org/10.3390/ijms20246195
Chicago/Turabian StyleBascos, Neil Andrew D., and Samuel J. Landry. 2019. "A History of Molecular Chaperone Structures in the Protein Data Bank" International Journal of Molecular Sciences 20, no. 24: 6195. https://doi.org/10.3390/ijms20246195
APA StyleBascos, N. A. D., & Landry, S. J. (2019). A History of Molecular Chaperone Structures in the Protein Data Bank. International Journal of Molecular Sciences, 20(24), 6195. https://doi.org/10.3390/ijms20246195