AMPK-Mediated Regulation of Alpha-Arrestins and Protein Trafficking


Abstract
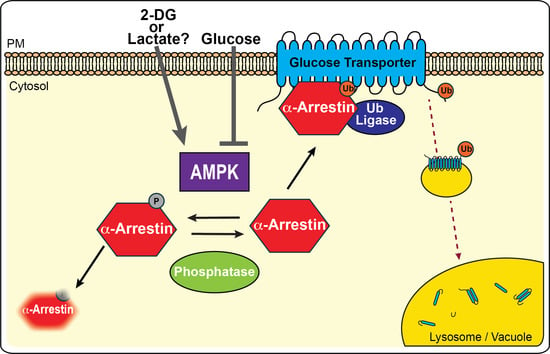
:
1. Introduction
2. The Arrestin Family of Protein Trafficking Adaptors
3. AMPK-Regulation of α-Arrestin-Mediated Trafficking in S. cerevisiae
3.1. AMPK Inhibits α-Arrestin-Mediated Trafficking of Glucose Transporters
3.2. AMPK Inhibits α-Arrestin-Mediated Trafficking of the Jen1 Lactate Permease and Other Membrane Cargo
4. AMPK- and Carbon-Source Regulation of α-Arrestin Gene Expression
4.1. Control of Txnip Expression by MondoA and ChREBP
4.2. Control of ArrDC4 Expression by MondoA
4.3. Altered Expression of Yeast α-Arrestins in Response to Carbon Supply
5. AMPK Regulation of α-Arrestin-Mediated Trafficking in Mammals
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AICAR | 5-Aminoimidazole-4-carboxamide ribonucleotide |
| AMP | Adenosine Monophosphate |
| ADP | Adenosine Diphosphate |
| ARRDC | Arrestin Domain Containing Protein |
| ATP | Adenosine Triphosphate |
| GLUT | Glucose Transporters |
| GPCR | G-protein coupled receptor |
| HXT | Hexose Transporters |
| NEDD4 | Neural precursor cell Expressed, Developmentally Down-regulated 4 |
| PP2A | Protein phosphatase 2A |
| RAB-GAP | RAB-GTPase activating protein |
| Rsp5 | Reverses Spt- Phenotype 5 |
| TBC1D | TBC1 domain family member 1D |
| TXNIP | Thioredoxin Interacting Protein |
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| LD | linear dichroism |
References
- Deng, D.; Yan, N. GLUT, SGLT, and SWEET: Structural and mechanistic investigations of the glucose transporters. Protein Sci. Publ. Protein Soc. 2016, 25, 546–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, S.; Johnston, M. Function and regulation of yeast hexose transporters. Microbiol. Mol. Biol. Rev. 1999, 63, 554–569. [Google Scholar] [PubMed]
- Augustin, R. The protein family of glucose transport facilitators: It’s not only about glucose after all. IUBMB Life 2010, 62, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Carling, D.; Carlson, M. The AMP-activated/SNF1 protein kinase subfamily: Metabolic sensors of the eukaryotic cell? Annu. Rev. Biochem. 1998, 67, 821–855. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.R., 3rd; Bergeron, R.; Shulman, G.I.; Young, L.H. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am. J. Physiol. 1999, 277, H643–H649. [Google Scholar] [CrossRef] [PubMed]
- Kurth-Kraczek, E.J.; Hirshman, M.F.; Goodyear, L.J.; Winder, W.W. 5’ AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes 1999, 48, 1667–1671. [Google Scholar] [CrossRef] [PubMed]
- Fryer, L.G.; Foufelle, F.; Barnes, K.; Baldwin, S.A.; Woods, A.; Carling, D. Characterization of the role of the AMP-activated protein kinase in the stimulation of glucose transport in skeletal muscle cells. Biochem. J. 2002, 363, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Barnes, K.; Ingram, J.C.; Porras, O.H.; Barros, L.F.; Hudson, E.R.; Fryer, L.G.; Foufelle, F.; Carling, D.; Hardie, D.G.; Baldwin, S.A. Activation of GLUT1 by metabolic and osmotic stress: Potential involvement of AMP-activated protein kinase (AMPK). J. Cell Sci. 2002, 115, 2433–2442. [Google Scholar]
- Abbud, W.; Habinowski, S.; Zhang, J.Z.; Kendrew, J.; Elkairi, F.S.; Kemp, B.E.; Witters, L.A.; Ismail-Beigi, F. Stimulation of AMP-activated protein kinase (AMPK) is associated with enhancement of Glut1-mediated glucose transport. Arch. Biochem. Biophys. 2000, 380, 347–352. [Google Scholar] [CrossRef]
- Chen, S.; Murphy, J.; Toth, R.; Campbell, D.G.; Morrice, N.A.; Mackintosh, C. Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. Biochem. J. 2008, 409, 449–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pehmoller, C.; Treebak, J.T.; Birk, J.B.; Chen, S.; Mackintosh, C.; Hardie, D.G.; Richter, E.A.; Wojtaszewski, J.F. Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am. J. Physiol. Endocrinol. MeTable 2009, 297, E665–E675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, S.A.; Barros, L.F.; Griffiths, M.; Ingram, J.; Robbins, E.C.; Streets, A.J.; Saklatvala, J. Regulation of GLUT1 in response to cellular stress. Biochem. Soc. Trans. 1997, 25, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Waldhart, A.N.; Dykstra, H.; Peck, A.S.; Boguslawski, E.A.; Madaj, Z.B.; Wen, J.; Veldkamp, K.; Hollowell, M.; Zheng, B.; Cantley, L.C.; et al. Phosphorylation of TXNIP by AKT Mediates Acute Influx of Glucose in Response to Insulin. Cell Rep. 2017, 19, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Patwari, P.; Lee, R.T. An expanded family of arrestins regulate metabolism. Trends Endocrinol. Metab. TEM 2012, 23, 216–222. [Google Scholar] [CrossRef] [Green Version]
- Becuwe, M.; Vieira, N.; Lara, D.; Gomes-Rezende, J.; Soares-Cunha, C.; Casal, M.; Haguenauer-Tsapis, R.; Vincent, O.; Paiva, S.; Leon, S. A molecular switch on an arrestin-like protein relays glucose signaling to transporter endocytosis. J. Cell Biol. 2012, 196, 247–259. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; MacGurn, J.A.; Chu, T.; Stefan, C.J.; Emr, S.D. Arrestin-related ubiquitin-ligase adaptors regulate endocytosis and protein turnover at the cell surface. Cell 2008, 135, 714–725. [Google Scholar] [CrossRef]
- Nikko, E.; Pelham, H.R. Arrestin-mediated endocytosis of yeast plasma membrane transporters. Traffic 2009, 10, 1856–1867. [Google Scholar] [CrossRef]
- O’Donnell, A.F.; Apffel, A.; Gardner, R.G.; Cyert, M.S. Alpha-arrestins Aly1 and Aly2 regulate intracellular trafficking in response to nutrient signaling. Mol. Biol. Cell 2010, 21, 3552–3566. [Google Scholar] [CrossRef]
- Nikko, E.; Sullivan, J.A.; Pelham, H.R. Arrestin-like proteins mediate ubiquitination and endocytosis of the yeast metal transporter Smf1. EMBO Rep. 2008, 9, 1216–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, A.F.; McCartney, R.R.; Chandrashekarappa, D.G.; Zhang, B.B.; Thorner, J.; Schmidt, M.C. 2-Deoxyglucose impairs Saccharomyces cerevisiae growth by stimulating Snf1-regulated and alpha-arrestin-mediated trafficking of hexose transporters 1 and 3. Mol. Cell. Biol. 2015, 35, 939–955. [Google Scholar] [CrossRef] [PubMed]
- Cha-Molstad, H.; Saxena, G.; Chen, J.; Shalev, A. Glucose-stimulated expression of Txnip is mediated by carbohydrate response element-binding protein, p300, and histone H4 acetylation in pancreatic beta cells. J. Biol. Chem. 2009, 284, 16898–16905. [Google Scholar] [CrossRef] [PubMed]
- Hovsepian, J.; Defenouillere, Q.; Albanese, V.; Vachova, L.; Garcia, C.; Palkova, Z.; Leon, S. Multilevel regulation of an alpha-arrestin by glucose depletion controls hexose transporter endocytosis. J. Cell Biol. 2017, 216, 1811–1831. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Osatomi, K.; Yamashita, H.; Kabashima, T.; Uyeda, K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: Regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J. Biol. Chem. 2002, 277, 3829–3835. [Google Scholar] [CrossRef] [PubMed]
- Shalev, A.; Pise-Masison, C.A.; Radonovich, M.; Hoffmann, S.C.; Hirshberg, B.; Brady, J.N.; Harlan, D.M. Oligonucleotide microarray analysis of intact human pancreatic islets: Identification of glucose-responsive genes and a highly regulated TGFbeta signaling pathway. Endocrinology 2002, 143, 3695–3698. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.E. On the origins of arrestin and rhodopsin. BMC Evol. Biol. 2008, 8, 222. [Google Scholar] [CrossRef]
- Shi, H.; Rojas, R.; Bonifacino, J.S.; Hurley, J.H. The retromer subunit Vps26 has an arrestin fold and binds Vps35 through its C-terminal domain. Nat. Struct. Mol. Biol. 2006, 13, 540–548. [Google Scholar] [CrossRef]
- Granzin, J.; Cousin, A.; Weirauch, M.; Schlesinger, R.; Buldt, G.; Batra-Safferling, R. Crystal structure of p44, a constitutively active splice variant of visual arrestin. J. Mol. Biol. 2012, 416, 611–618. [Google Scholar] [CrossRef]
- Han, M.; Gurevich, V.V.; Vishnivetskiy, S.A.; Sigler, P.B.; Schubert, C. Crystal structure of beta-arrestin at 1.9 A: Possible mechanism of receptor binding and membrane Translocation. Structure (Lond.) 2001, 9, 869–880. [Google Scholar] [CrossRef]
- Hwang, J.; Suh, H.W.; Jeon, Y.H.; Hwang, E.; Nguyen, L.T.; Yeom, J.; Lee, S.G.; Lee, C.; Kim, K.J.; Kang, B.S.; et al. The structural basis for the negative regulation of thioredoxin by thioredoxin-interacting protein. Nat. Commun. 2014, 5, 2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, Z.Y.; Puthenveedu, M.A. Regulation of G protein-coupled receptor signaling by plasma membrane organization and endocytosis. Traffic 2018. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.E.; Lefkowitz, R.J. Expanding roles for beta-arrestins as scaffolds and adapters in GPCR signaling and trafficking. Curr. Opin. Cell Biol. 2001, 13, 139–145. [Google Scholar] [CrossRef]
- Jean-Charles, P.Y.; Kaur, S.; Shenoy, S.K. G Protein-Coupled Receptor Signaling Through beta-Arrestin-Dependent Mechanisms. J. Cardiovasc. Pharmacol. 2017, 70, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Shenoy, S.K. GPCR desensitization: Acute and prolonged phases. Cell Signal. 2018, 41, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; DeFea, K.A. beta-Arrestin-kinase scaffolds: Turn them on or turn them off? Wiley Interdiscip. Rev. Syst. Biol. Med. 2013, 5, 231–241. [Google Scholar] [CrossRef]
- MacGurn, J.A.; Hsu, P.C.; Smolka, M.B.; Emr, S.D. TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell 2011, 147, 1104–1117. [Google Scholar] [CrossRef]
- Han, S.O.; Kommaddi, R.P.; Shenoy, S.K. Distinct roles for beta-arrestin2 and arrestin-domain-containing proteins in beta2 adrenergic receptor trafficking. EMBO Rep. 2013, 14, 164–171. [Google Scholar] [CrossRef]
- Nabhan, J.F.; Pan, H.; Lu, Q. Arrestin domain-containing protein 3 recruits the NEDD4 E3 ligase to mediate ubiquitination of the beta2-adrenergic receptor. EMBO Rep. 2010, 11, 605–611. [Google Scholar] [CrossRef]
- Alvaro, C.G.; Aindow, A.; Thorner, J. Differential Phosphorylation Provides a Switch to Control How alpha-Arrestin Rod1 Down-regulates Mating Pheromone Response in Saccharomyces cerevisiae. Genetics 2016, 203, 299–317. [Google Scholar] [CrossRef]
- Prosser, D.C.; Pannunzio, A.E.; Brodsky, J.L.; Thorner, J.; Wendland, B.; O’Donnell, A.F. alpha-Arrestins participate in cargo selection for both clathrin-independent and clathrin-mediated endocytosis. J. Cell Sci. 2015, 128, 4220–4234. [Google Scholar] [CrossRef] [PubMed]
- Smardon, A.M.; Kane, P.M. Loss of vacuolar H+-ATPase activity in organelles signals ubiquitination and endocytosis of the yeast plasma membrane proton pump Pma1p. J. Biol. Chem. 2014, 289, 32316–32326. [Google Scholar] [CrossRef]
- O’Donnell, A.F.; Huang, L.; Thorner, J.; Cyert, M.S. A calcineurin-dependent switch controls the trafficking function of alpha-arrestin Aly1/Art6. J. Biol. Chem. 2013, 288, 24063–24080. [Google Scholar] [CrossRef] [PubMed]
- Crapeau, M.; Merhi, A.; Andre, B. Stress conditions promote yeast Gap1 permease ubiquitylation and down-regulation via the arrestin-like Bul and Aly proteins. J. Biol. Chem. 2014, 289, 22103–22116. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, R.; Kamiya, M.; Takahara, T.; Maeda, T. Endocytosis of the aspartic acid/glutamic acid transporter Dip5 is triggered by substrate-dependent recruitment of the Rsp5 ubiquitin ligase via the arrestin-like protein Aly2. Mol. Cell. Biol. 2010, 30, 5598–5607. [Google Scholar] [CrossRef]
- Hovsepian, J.; Albanese, V.; Becuwe, M.; Ivashov, V.; Teis, D.; Leon, S. The yeast arrestin-related protein Bul1 is a novel actor of glucose-induced endocytosis. Mol. Biol. Cell 2018, 29, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Moriya, A.; Uemura, S.; Abe, F. Functional implications and ubiquitin-dependent degradation of the peptide transporter Ptr2 in Saccharomyces cerevisiae. Eukaryot. Cell 2014, 13, 1380–1392. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Mochizuki, T.; Uemura, S.; Hiraki, T.; Abe, F. Pressure-induced endocytic degradation of the Saccharomyces cerevisiae low-affinity tryptophan permease Tat1 is mediated by Rsp5 ubiquitin ligase and functionally redundant PPxY motif proteins. Eukaryot. Cell 2013, 12, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Merhi, A.; Andre, B. Internal amino acids promote Gap1 permease ubiquitylation via TORC1/Npr1/14-3-3-dependent control of the Bul arrestin-like adaptors. Mol. Cell. Biol. 2012, 32, 4510–4522. [Google Scholar] [CrossRef]
- Liu, J.; Sitaram, A.; Burd, C.G. Regulation of copper-dependent endocytosis and vacuolar degradation of the yeast copper transporter, Ctr1p, by the Rsp5 ubiquitin ligase. Traffic 2007, 8, 1375–1384. [Google Scholar] [CrossRef]
- Abe, F.; Iida, H. Pressure-induced differential regulation of the two tryptophan permeases Tat1 and Tat2 by ubiquitin ligase Rsp5 and its binding proteins, Bul1 and Bul2. Mol. Cell. Biol. 2003, 23, 7566–7584. [Google Scholar] [CrossRef] [PubMed]
- Villers, J.; Savocco, J.; Szopinska, A.; Degand, H.; Nootens, S.; Morsomme, P. Study of the Plasma Membrane Proteome Dynamics Reveals Novel Targets of the Nitrogen Regulation in Yeast. Mol. Cell. Proteom. 2017, 16, 1652–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snowdon, C.; van der Merwe, G. Regulation of Hxt3 and Hxt7 turnover converges on the Vid30 complex and requires inactivation of the Ras/cAMP/PKA pathway in Saccharomyces cerevisiae. PLoS ONE 2012, 7, e50458. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, C.G.; O’Donnell, A.F.; Prosser, D.C.; Augustine, A.A.; Goldman, A.; Brodsky, J.L.; Cyert, M.S.; Wendland, B.; Thorner, J. Specific alpha-arrestins negatively regulate Saccharomyces cerevisiae pheromone response by down-modulating the G-protein coupled receptor Ste2. Mol. Cell. Biol. 2014. [Google Scholar] [CrossRef]
- Herrador, A.; Livas, D.; Soletto, L.; Becuwe, M.; Leon, S.; Vincent, O. Casein kinase 1 controls the activation threshold of an alpha-arrestin by multisite phosphorylation of the interdomain hinge. Mol. Biol. Cell 2015, 26, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Raja, J.; Davis, D.A. The beta-arrestin-like protein Rim8 is hyperphosphorylated and complexes with Rim21 and Rim101 to promote adaptation to neutral-alkaline pH. Eukaryot. Cell 2012, 11, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Shi, Q.; Li, W.; Mu, X.; Peng, J.; Li, M.; Chen, M.; Huang, H.; Wang, C.; Gao, K.; et al. ARRDC1 and ARRDC3 act as tumor suppressors in renal cell carcinoma by facilitating YAP1 degradation. Am. J. Cancer Res. 2018, 8, 132–143. [Google Scholar]
- Puca, L.; Chastagner, P.; Meas-Yedid, V.; Israel, A.; Brou, C. Alpha-arrestin 1 (ARRDC1) and beta-arrestins cooperate to mediate Notch degradation in mammals. J. Cell Sci. 2013, 126, 4457–4468. [Google Scholar] [CrossRef]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, K.; Foot, N.J.; Anand, S.; Dalton, H.E.; Chaudhary, N.; Collins, B.M.; Mathivanan, S.; Kumar, S. Regulation of the divalent metal ion transporter via membrane budding. Cell Discov. 2016, 2, 16011. [Google Scholar] [CrossRef] [Green Version]
- Dores, M.R.; Lin, H.; Grimsey, N.J.; Mendez, F.; Trejo, J. The alpha-arrestin ARRDC3 mediates ALIX ubiquitination and G protein-coupled receptor lysosomal sorting. Mol. Biol. Cell 2015, 26, 4660–4673. [Google Scholar] [CrossRef]
- Shea, F.F.; Rowell, J.L.; Li, Y.; Chang, T.H.; Alvarez, C.E. Mammalian alpha arrestins link activated seven transmembrane receptors to Nedd4 family e3 ubiquitin ligases and interact with beta arrestins. PLoS ONE 2012, 7, e50557. [Google Scholar] [CrossRef]
- Soung, Y.H.; Ford, S.; Yan, C.; Chung, J. The Role of Arrestin Domain-Containing 3 in Regulating Endocytic Recycling and Extracellular Vesicle Sorting of Integrin beta4 in Breast Cancer. Cancers 2018, 10, 507. [Google Scholar]
- Hager, N.A.; Krasowski, C.J.; Mackie, T.D.; Kolb, A.R.; Needham, P.G.; Augustine, A.A.; Dempsey, A.; Szent-Gyorgyi, C.; Bruchez, M.P.; Bain, D.J.; et al. Select alpha-arrestins control cell-surface abundance of the mammalian Kir2.1 potassium channel in a yeast model. J. Biol. Chem. 2018, 293, 11006–11021. [Google Scholar] [CrossRef] [PubMed]
- Llopis-Torregrosa, V.; Ferri-Blazquez, A.; Adam-Artigues, A.; Deffontaines, E.; van Heusden, G.P.; Yenush, L. Regulation of the Yeast Hxt6 Hexose Transporter by the Rod1 alpha-Arrestin, the Snf1 Protein Kinase, and the Bmh2 14-3-3 Protein. J. Biol. Chem. 2016, 291, 14973–14985. [Google Scholar] [CrossRef] [PubMed]
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villen, J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 2013, 10, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Holt, L.J.; Tuch, B.B.; Villen, J.; Johnson, A.D.; Gygi, S.P.; Morgan, D.O. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 2009, 325, 1682–1686. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, C.P.; Smolka, M.B.; Payne, S.H.; Bafna, V.; Eng, J.; Zhou, H. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol. Cell. Proteom. 2008, 7, 1389–1396. [Google Scholar] [CrossRef]
- Muir, A.; Ramachandran, S.; Roelants, F.M.; Timmons, G.; Thorner, J. TORC2-dependent protein kinase Ypk1 phosphorylates ceramide synthase to stimulate synthesis of complex sphingolipids. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.Z.; Ao, S.Z. Analysis of phosphorylation of YJL084c, a yeast protein. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao Acta Biochim. Biophys. Sin. 2002, 34, 433–438. [Google Scholar]
- Ptacek, J.; Devgan, G.; Michaud, G.; Zhu, H.; Zhu, X.; Fasolo, J.; Guo, H.; Jona, G.; Breitkreutz, A.; Sopko, R.; et al. Global analysis of protein phosphorylation in yeast. Nature 2005, 438, 679–684. [Google Scholar] [CrossRef] [Green Version]
- Papinski, D.; Kraft, C. Atg1 kinase organizes autophagosome formation by phosphorylating Atg9. Autophagy 2014, 10, 1338–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, A.; Roy, J.; Bodenmiller, B.; Wanka, S.; Landry, C.R.; Aebersold, R.; Cyert, M.S. The calcineurin signaling network evolves via conserved kinase-phosphatase modules that transcend substrate identity. Mol. Cell 2014, 55, 422–435. [Google Scholar] [CrossRef] [PubMed]
- Talaia, G.; Gournas, C.; Saliba, E.; Barata-Antunes, C.; Casal, M.; Andre, B.; Diallinas, G.; Paiva, S. The alpha-Arrestin Bul1p Mediates Lactate Transporter Endocytosis in Response to Alkalinization and Distinct Physiological Signals. J. Mol. Biol. 2017, 429, 3678–3695. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, A.F. The running of the Buls: Control of permease trafficking by alpha-arrestins Bul1 and Bul2. Mol. Cell. Biol. 2012, 32, 4506–4509. [Google Scholar] [CrossRef] [PubMed]
- Becuwe, M.; Leon, S. Integrated control of transporter endocytosis and recycling by the arrestin-related protein Rod1 and the ubiquitin ligase Rsp5. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.C.; McCartney, R.R. beta-subunits of Snf1 kinase are required for kinase function and substrate definition. EMBO J. 2000, 19, 4936–4943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedbacker, K.; Carlson, M. Regulation of the nucleocytoplasmic distribution of Snf1-Gal83 protein kinase. Eukaryot. Cell 2006, 5, 1950–1956. [Google Scholar] [CrossRef]
- Vincent, O.; Townley, R.; Kuchin, S.; Carlson, M. Subcellular localization of the Snf1 kinase is regulated by specific beta subunits and a novel glucose signaling mechanism. Genes Dev. 2001, 15, 1104–1114. [Google Scholar] [CrossRef] [Green Version]
- McCartney, R.R.; Schmidt, M.C. Regulation of Snf1 kinase. Activation requires phosphorylation of threonine 210 by an upstream kinase as well as a distinct step mediated by the Snf4 subunit. J. Biol. Chem. 2001, 276, 36460–36466. [Google Scholar] [CrossRef]
- McCartney, R.R.; Rubenstein, E.M.; Schmidt, M.C. Snf1 kinase complexes with different beta subunits display stress-dependent preferences for the three Snf1-activating kinases. Curr. Genet. 2005, 47, 335–344. [Google Scholar] [CrossRef]
- Hong, S.P.; Carlson, M. Regulation of snf1 protein kinase in response to environmental stress. J. Biol. Chem. 2007, 282, 16838–16845. [Google Scholar] [CrossRef] [PubMed]
- Hiesinger, M.; Roth, S.; Meissner, E.; Schuller, H.J. Contribution of Cat8 and Sip4 to the transcriptional activation of yeast gluconeogenic genes by carbon source-responsive elements. Curr. Genet. 2001, 39, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Vincent, O.; Carlson, M. Sip4, a Snf1 kinase-dependent transcriptional activator, binds to the carbon source-responsive element of gluconeogenic genes. EMBO J. 1998, 17, 7002–7008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, E.T.; Dombek, K.M.; Tachibana, C.; Ideker, T. Multiple pathways are co-regulated by the protein kinase Snf1 and the transcription factors Adr1 and Cat8. J. Biol. Chem. 2003, 278, 26146–26158. [Google Scholar] [CrossRef] [PubMed]
- Ostling, J.; Ronne, H. Negative control of the Mig1p repressor by Snf1p-dependent phosphorylation in the absence of glucose. Eur. J. Biochem. 1998, 252, 162–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, F.C.; Davies, S.P.; Wilson, W.A.; Carling, D.; Hardie, D.G. The SNF1 kinase complex from Saccharomyces cerevisiae phosphorylates the transcriptional repressor protein Mig1p in vitro at four sites within or near regulatory domain 1. FEBS Lett. 1999, 453, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Treitel, M.A.; Kuchin, S.; Carlson, M. Snf1 protein kinase regulates phosphorylation of the Mig1 repressor in Saccharomyces cerevisiae. Mol. Cell. Biol. 1998, 18, 6273–6280. [Google Scholar] [CrossRef] [PubMed]
- Serra-Cardona, A.; Petrezselyova, S.; Canadell, D.; Ramos, J.; Arino, J. Coregulated expression of the Na+/phosphate Pho89 transporter and Ena1 Na+-ATPase allows their functional coupling under high-pH stress. Mol. Cell. Biol. 2014, 34, 4420–4435. [Google Scholar] [CrossRef]
- Chandrashekarappa, D.G.; McCartney, R.R.; O’Donnell, A.F.; Schmidt, M.C. The beta subunit of yeast AMP-activated protein kinase directs substrate specificity in response to alkaline stress. Cell Signal. 2016, 28, 1881–1893. [Google Scholar] [CrossRef]
- Zaman, S.; Lippman, S.I.; Schneper, L.; Slonim, N.; Broach, J.R. Glucose regulates transcription in yeast through a network of signaling pathways. Mol. Syst. Biol. 2009, 5, 245. [Google Scholar] [CrossRef]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.; Park, S.; Kim, M.J.; Yang, W.K.; Im, D.U.; Yang, K.R.; Hong, J.; Choe, W.; Kang, I.; Kim, S.S.; et al. AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS J. 2014, 281, 4421–4438. [Google Scholar] [CrossRef] [Green Version]
- Shinoda, J.; Kikuchi, Y. Rod1, an arrestin-related protein, is phosphorylated by Snf1-kinase in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2007, 364, 258–263. [Google Scholar] [CrossRef] [PubMed]
- McCartney, R.R.; Chandrashekarappa, D.G.; Zhang, B.B.; Schmidt, M.C. Genetic analysis of resistance and sensitivity to 2-deoxyglucose in Saccharomyces cerevisiae. Genetics 2014, 198, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Casal, M.; Queiros, O.; Talaia, G.; Ribas, D.; Paiva, S. Carboxylic acids plasma membrane transporters in Saccharomyces cerevisiae. Adv. Exp. Med. Biol. 2016, 892, 229–251. [Google Scholar] [PubMed]
- Bozaquel-Morais, B.L.; Madeira, J.B.; Maya-Monteiro, C.M.; Masuda, C.A.; Montero-Lomeli, M. A new fluorescence-based method identifies protein phosphatases regulating lipid droplet metabolism. PLoS ONE 2010, 5, e13692. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Liu, Y.; Xu, X.; Carlson, M. Heterotrimer-independent regulation of activation-loop phosphorylation of Snf1 protein kinase involves two protein phosphatases. Proc. Natl. Acad. Sci. USA 2012, 109, 8652–8657. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Pei, G. Arrestins in metabolic regulation. Prog. Mol. Biol. Transl. Sci. 2013, 118, 413–427. [Google Scholar]
- Hupfeld, C.J.; Olefsky, J.M. Regulation of receptor tyrosine kinase signaling by GRKs and beta-arrestins. Annu. Rev. Physiol. 2007, 69, 561–577. [Google Scholar] [CrossRef]
- Luan, B.; Zhao, J.; Wu, H.; Duan, B.; Shu, G.; Wang, X.; Li, D.; Jia, W.; Kang, J.; Pei, G. Deficiency of a beta-arrestin-2 signal complex contributes to insulin resistance. Nature 2009, 457, 1146–1149. [Google Scholar] [CrossRef]
- Patwari, P.; Emilsson, V.; Schadt, E.E.; Chutkow, W.A.; Lee, S.; Marsili, A.; Zhang, Y.; Dobrin, R.; Cohen, D.E.; Larsen, P.R.; et al. The arrestin domain-containing 3 protein regulates body mass and energy expenditure. Cell MeTable 2011, 14, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Patwari, P.; Chutkow, W.A.; Cummings, K.; Verstraeten, V.L.; Lammerding, J.; Schreiter, E.R.; Lee, R.T. Thioredoxin-independent regulation of metabolism by the alpha-arrestin proteins. J. Biol. Chem. 2009, 284, 24996–25003. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, J.S.; Chatterjee, A.; Castellani, L.W.; Ross, D.A.; Ohmen, J.; Cavalcoli, J.; Wu, C.; Dains, K.M.; Catanese, J.; Chu, M.; et al. Positional cloning of the combined hyperlipidemia gene Hyplip1. Nat. Genet. 2002, 30, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Stoltzman, C.A.; Peterson, C.W.; Breen, K.T.; Muoio, D.M.; Billin, A.N.; Ayer, D.E. Glucose sensing by MondoA:Mlx complexes: A role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6912–6917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kibbe, C.; Chen, J.; Xu, G.; Jing, G.; Shalev, A. FOXO1 competes with carbohydrate response element-binding protein (ChREBP) and inhibits thioredoxin-interacting protein (TXNIP) transcription in pancreatic beta cells. J. Biol. Chem. 2013, 288, 23194–23202. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hui, S.T.; Couto, F.M.; Mungrue, I.N.; Davis, D.B.; Attie, A.D.; Lusis, A.J.; Davis, R.A.; Shalev, A. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 3581–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minn, A.H.; Hafele, C.; Shalev, A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology 2005, 146, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Richards, P.; Rachdi, L.; Oshima, M.; Marchetti, P.; Bugliani, M.; Armanet, M.; Postic, C.; Guilmeau, S.; Scharfmann, R. MondoA Is an Essential Glucose-Responsive Transcription Factor in Human Pancreatic beta-Cells. Diabetes 2018, 67, 461–472. [Google Scholar] [CrossRef]
- Patwari, P.; Higgins, L.J.; Chutkow, W.A.; Yoshioka, J.; Lee, R.T. The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange. J. Biol. Chem. 2006, 281, 21884–21891. [Google Scholar] [CrossRef]
- Oka, S.; Masutani, H.; Liu, W.; Horita, H.; Wang, D.; Kizaka-Kondoh, S.; Yodoi, J. Thioredoxin-binding protein-2-like inducible membrane protein is a novel vitamin D3 and peroxisome proliferator-activated receptor (PPAR)gamma ligand target protein that regulates PPARgamma signaling. Endocrinology 2006, 147, 733–743. [Google Scholar] [CrossRef]
- Spindel, O.N.; World, C.; Berk, B.C. Thioredoxin interacting protein: Redox dependent and independent regulatory mechanisms. Antioxid. Redox Signal. 2012, 16, 587–596. [Google Scholar] [CrossRef]
- Havula, E.; Hietakangas, V. Sugar sensing by ChREBP/Mondo-Mlx-new insight into downstream regulatory networks and integration of nutrient-derived signals. Curr. Opin. Cell Biol. 2018, 51, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Richards, P.; Ourabah, S.; Montagne, J.; Burnol, A.F.; Postic, C.; Guilmeau, S. MondoA/ChREBP: The usual suspects of transcriptional glucose sensing; Implication in pathophysiology. Metab. Clin. Exp. 2017, 70, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.J.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Robinson, L.N.; Towle, H.C. ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J. Biol. Chem. 2006, 281, 28721–28730. [Google Scholar] [CrossRef]
- Stoltzman, C.A.; Kaadige, M.R.; Peterson, C.W.; Ayer, D.E. MondoA senses non-glucose sugars: Regulation of thioredoxin-interacting protein (TXNIP) and the hexose transport curb. J. Biol. Chem. 2011, 286, 38027–38034. [Google Scholar] [CrossRef] [PubMed]
- Sans, C.L.; Satterwhite, D.J.; Stoltzman, C.A.; Breen, K.T.; Ayer, D.E. MondoA-Mlx heterodimers are candidate sensors of cellular energy status: Mitochondrial localization and direct regulation of glycolysis. Mol. Cell. Biol. 2006, 26, 4863–4871. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715. [Google Scholar] [CrossRef] [Green Version]
- Svoboda, M.; Tastenoy, M.; Zhang, Y.; Gillet, C.; Rasschaert, J.; Malaisse, W.J.; Sener, A. D-glucose and 3-O-methyl-d-glucose-induced upregulation of selected genes in rat hepatocytes and INS1E cells: Reevaluation of the possible role of hexose phosphorylation. Mol. Med. Rep. 2013, 8, 829–836. [Google Scholar] [CrossRef]
- Dentin, R.; Tomas-Cobos, L.; Foufelle, F.; Leopold, J.; Girard, J.; Postic, C.; Ferre, P. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J. Hepatol. 2012, 56, 199–209. [Google Scholar] [CrossRef]
- Minn, A.H.; Couto, F.M.; Shalev, A. Metabolism-independent sugar effects on gene transcription: The role of 3-O-methylglucose. Biochemistry 2006, 45, 11047–11051. [Google Scholar] [CrossRef] [PubMed]
- Shaked, M.; Ketzinel-Gilad, M.; Cerasi, E.; Kaiser, N.; Leibowitz, G. AMP-activated protein kinase (AMPK) mediates nutrient regulation of thioredoxin-interacting protein (TXNIP) in pancreatic beta-cells. PLoS ONE 2011, 6, e28804. [Google Scholar] [CrossRef] [PubMed]
- Stoeckman, A.K.; Ma, L.; Towle, H.C. Mlx is the functional heteromeric partner of the carbohydrate response element-binding protein in glucose regulation of lipogenic enzyme genes. J. Biol. Chem. 2004, 279, 15662–15669. [Google Scholar] [CrossRef] [PubMed]
- Tsatsos, N.G.; Towle, H.C. Glucose activation of ChREBP in hepatocytes occurs via a two-step mechanism. Biochem. Biophys. Res. Commun. 2006, 340, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jing, G.; Xu, G.; Shalev, A. Thioredoxin-interacting protein stimulates its own expression via a positive feedback loop. Mol. Endocrinol. 2014, 28, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Baufeld, A.; Koczan, D.; Vanselow, J. Induction of altered gene expression profiles in cultured bovine granulosa cells at high cell density. Reprod. Biol. Endocrinol. 2017, 15, 3. [Google Scholar] [CrossRef]
- Robinson, K.A.; Koepke, J.I.; Kharodawala, M.; Lopes, J.M. A network of yeast basic helix-loop-helix interactions. Nucleic Acids Res. 2000, 28, 4460–4466. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.J.; Cole, M.D.; Erives, A.J. Evolution of the holozoan ribosome biogenesis regulon. BMC Genom. 2008, 9, 442. [Google Scholar] [CrossRef]
- McFerrin, L.G.; Atchley, W.R. Evolution of the Max and Mlx networks in animals. Genome Biol. Evol. 2011, 3, 915–937. [Google Scholar] [CrossRef]
- Merrill, G.F.; Kurth, E.J.; Hardie, D.G.; Winder, W.W. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am. J. Physiol. 1997, 273, E1107–E1112. [Google Scholar] [CrossRef] [PubMed]
- Daval, M.; Foufelle, F.; Ferre, P. Functions of AMP-activated protein kinase in adipose tissue. J. Physiol. 2006, 574, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.Y.; Li, H.; Pavlov, T.S.; Tuerk, R.D.; Tabares, D.; Brunisholz, R.; Neumann, D.; Staruschenko, A.; Hallows, K.R. beta1Pix exchange factor stabilizes the ubiquitin ligase Nedd4-2 and plays a critical role in ENaC regulation by AMPK in kidney epithelial cells. J. Biol. Chem. 2018, 293, 11612–11624. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Soler, N.M.; Hallows, K.R. AMP-activated protein kinase regulation of kidney tubular transport. Curr. Opin. Nephrol. Hypertens. 2012, 21, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Sakar, Y.; Meddah, B.; Faouzi, M.A.; Cherrah, Y.; Bado, A.; Ducroc, R. Metformin-induced regulation of the intestinal d-glucose transporters. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2010, 61, 301–307. [Google Scholar]
- Walker, J.; Jijon, H.B.; Diaz, H.; Salehi, P.; Churchill, T.; Madsen, K.L. 5-aminoimidazole-4-carboxamide riboside (AICAR) enhances GLUT2-dependent jejunal glucose transport: A possible role for AMPK. Biochem. J. 2005, 385, 485–491. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| α-Arrestin Genes and Cargos in Yeast | |||
|---|---|---|---|
| Gene | Alias | Cargo | References |
| ALY1 | ART6 | Gap1, Dip5, Ste3 | [20,41,43,44] |
| ALY2 | ART3 | Gap1, Dip5, Ste3 | [20,41,43,44,45] |
| ART5 | Itr1 | [19] | |
| ART10 | No known cargo | ||
| BUL1 | SMM2, DAG1, RDS1 | Jen1, Gap1, Ptr2, Tat1, Tat2, Ctr1, Put4, Dal5 | [46,47,48,49,50,51,52] |
| BUL2 | Gap1, Ptr2, Tat1, Tat2, Ctr1, Put4, Dal5 | [48,49,50,51,52] | |
| BUL3 | No known cargo | ||
| CSR2 | ART8, MRG19 | Hxt6, Hxt7, Hxt2, Hxt4 | [24,53] |
| ECM21 | ART2 | Tat2, Fur4, Lyp1, Smf1 | [19] |
| LDB19 | ART1 | Mup1, Ste2, Ste3, Can1, Lyp1, Tat2, Fur4 | [19,41,54] |
| RIM8 | ART9 | Rim21, Pma1 | [42,55,56] |
| ROD1 | ART4 | Hxt1, Hxt3, Hxt6, Jen1 | [17,19,22] |
| ROG3 | ART7 | Hxt3 | [22] |
| SPO23 | No known cargo | ||
| α-Arrestin Genes and Cargos in Humans | |||
|---|---|---|---|
| Gene | Alias | Cargo | References |
| ARRDC1 | YAP1, Notch, TSG101, DMT1 | [57,58,59,60] | |
| ARRDC2 | No known cargo | ||
| ARRDC3 | YAP1, PAR1, β3-AR, β2-AR, V2R, ITG β4 | [39,57,61,62,63] | |
| ARRDC4 | MDA5, DMT1, V2R, β2-AR | [60,62] | |
| ARRDC5 | No known cargo | ||
| TXNIP | VDUP1 | GLUT1, GLUT4 | [14,15] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Donnell, A.F.; Schmidt, M.C. AMPK-Mediated Regulation of Alpha-Arrestins and Protein Trafficking. Int. J. Mol. Sci. 2019, 20, 515. https://doi.org/10.3390/ijms20030515
O’Donnell AF, Schmidt MC. AMPK-Mediated Regulation of Alpha-Arrestins and Protein Trafficking. International Journal of Molecular Sciences. 2019; 20(3):515. https://doi.org/10.3390/ijms20030515
Chicago/Turabian StyleO’Donnell, Allyson F., and Martin C. Schmidt. 2019. "AMPK-Mediated Regulation of Alpha-Arrestins and Protein Trafficking" International Journal of Molecular Sciences 20, no. 3: 515. https://doi.org/10.3390/ijms20030515
APA StyleO’Donnell, A. F., & Schmidt, M. C. (2019). AMPK-Mediated Regulation of Alpha-Arrestins and Protein Trafficking. International Journal of Molecular Sciences, 20(3), 515. https://doi.org/10.3390/ijms20030515