Potent and Broad-Spectrum Antimicrobial Activity of Analogs from the Scorpion Peptide Stigmurin

 ,
,  , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. In Silico Analysis
2.2. Molecular Models
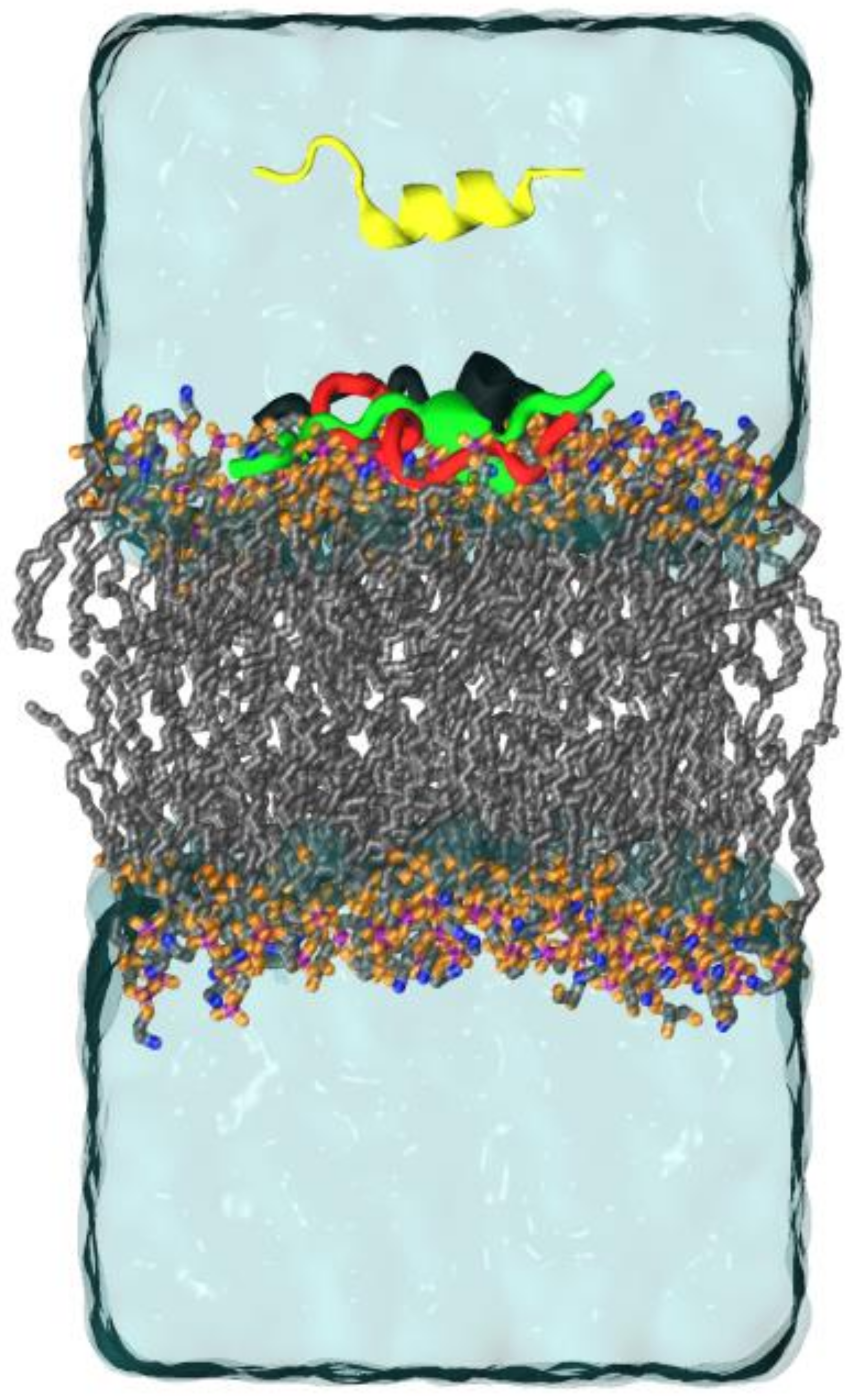
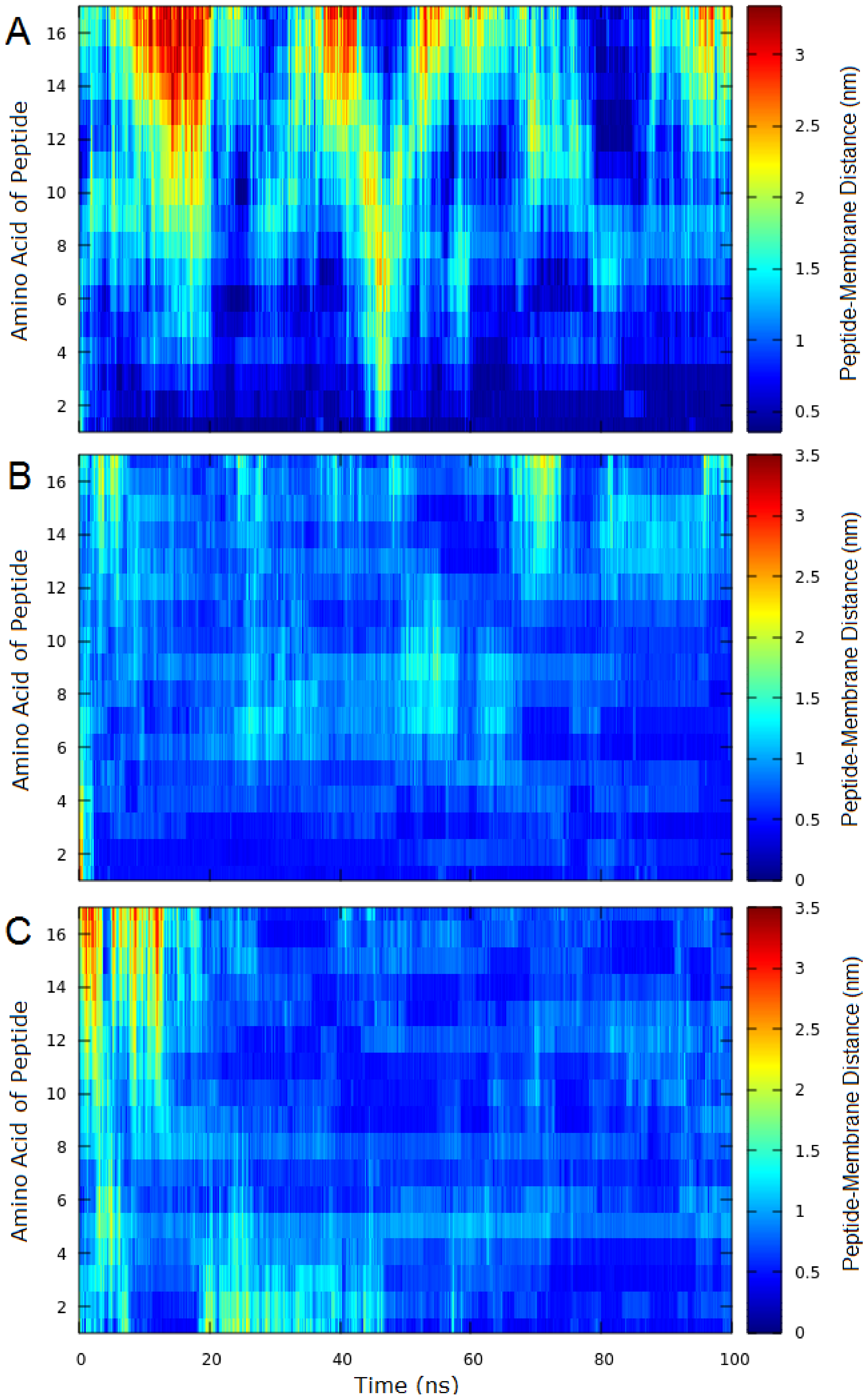
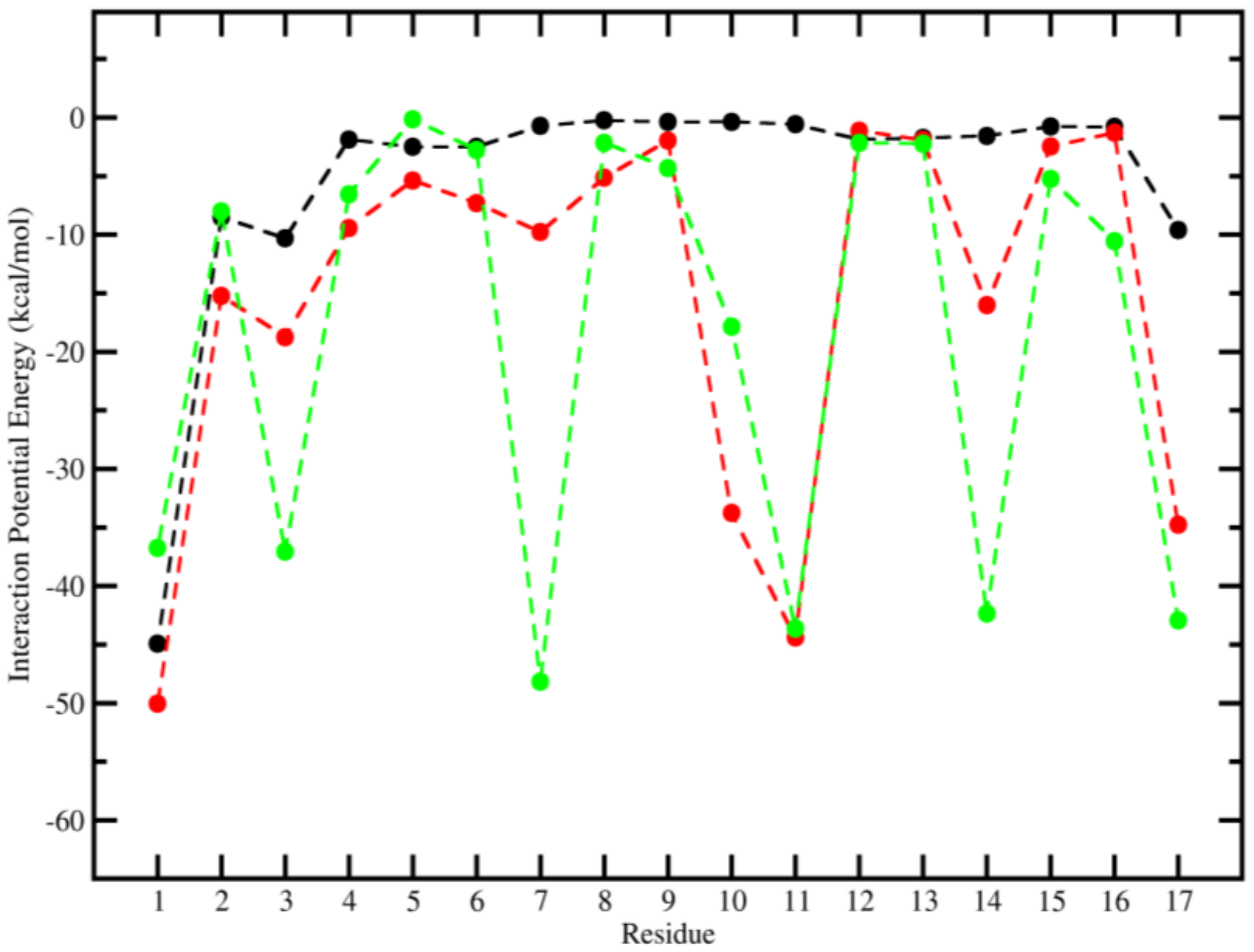
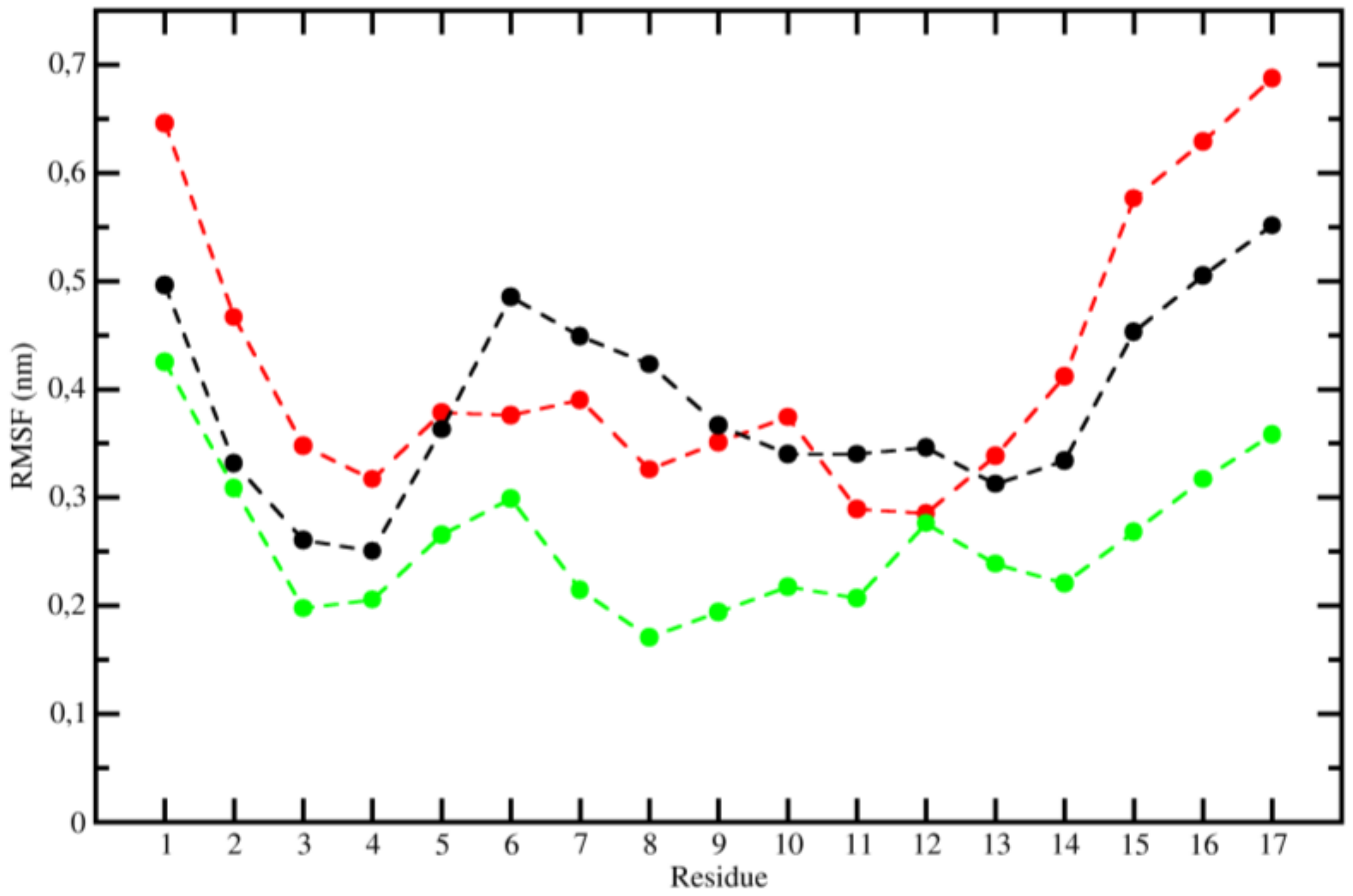
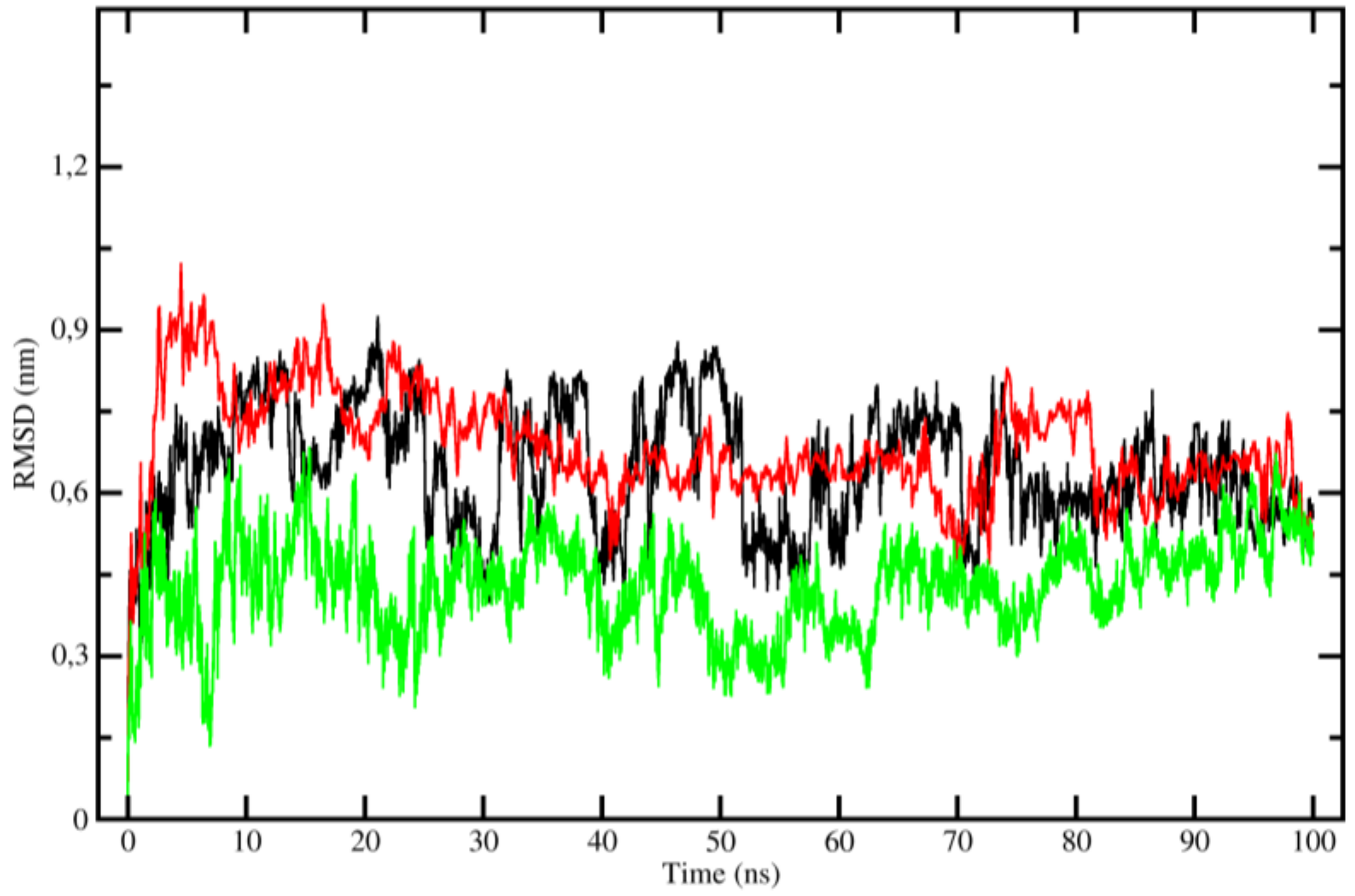
2.3. Molecular Dynamics (MD)
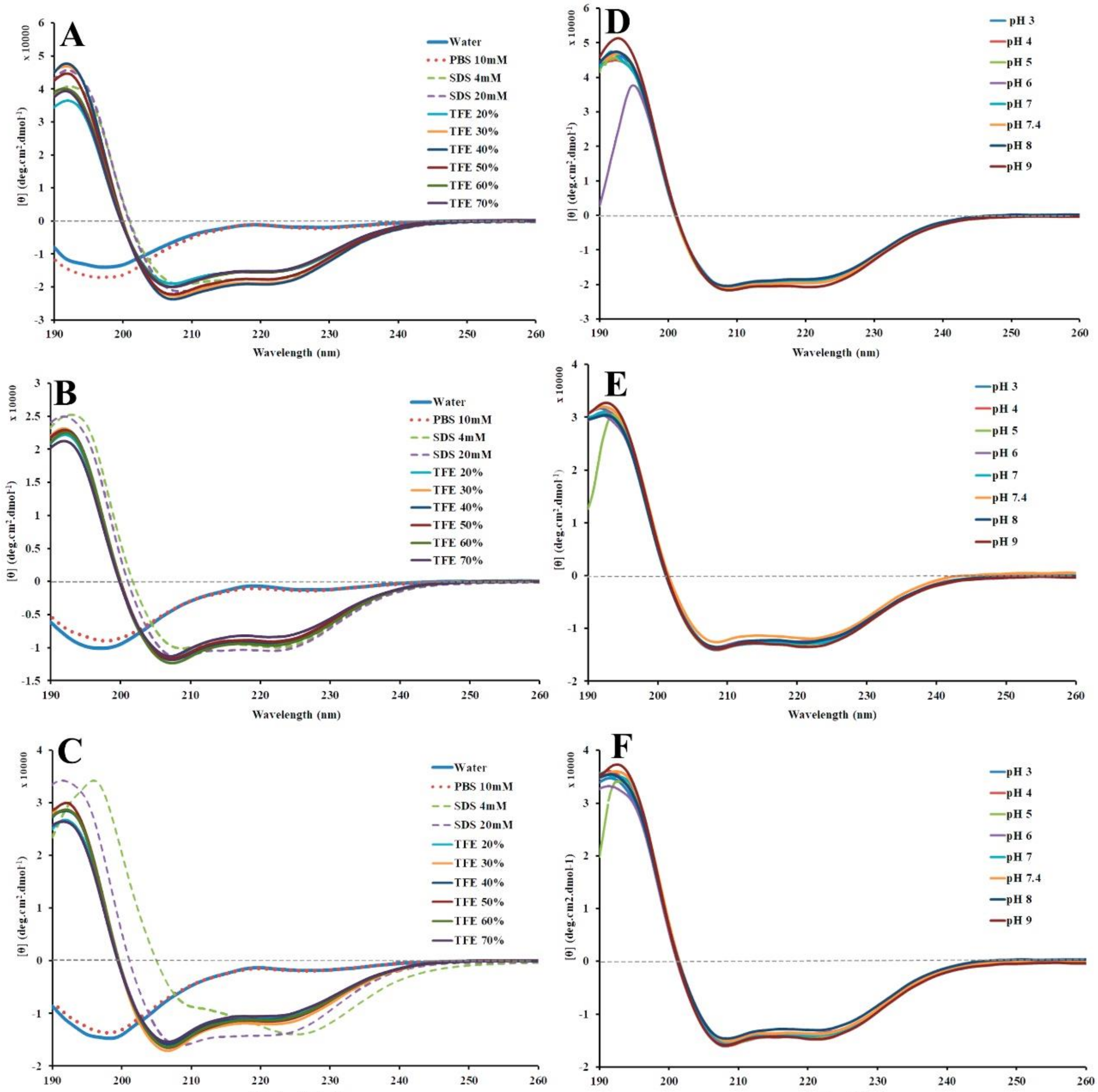
2.4. Structural Conformation by Circular Dichroism (CD)
2.5. Peptide Stability by Circular Dichroism
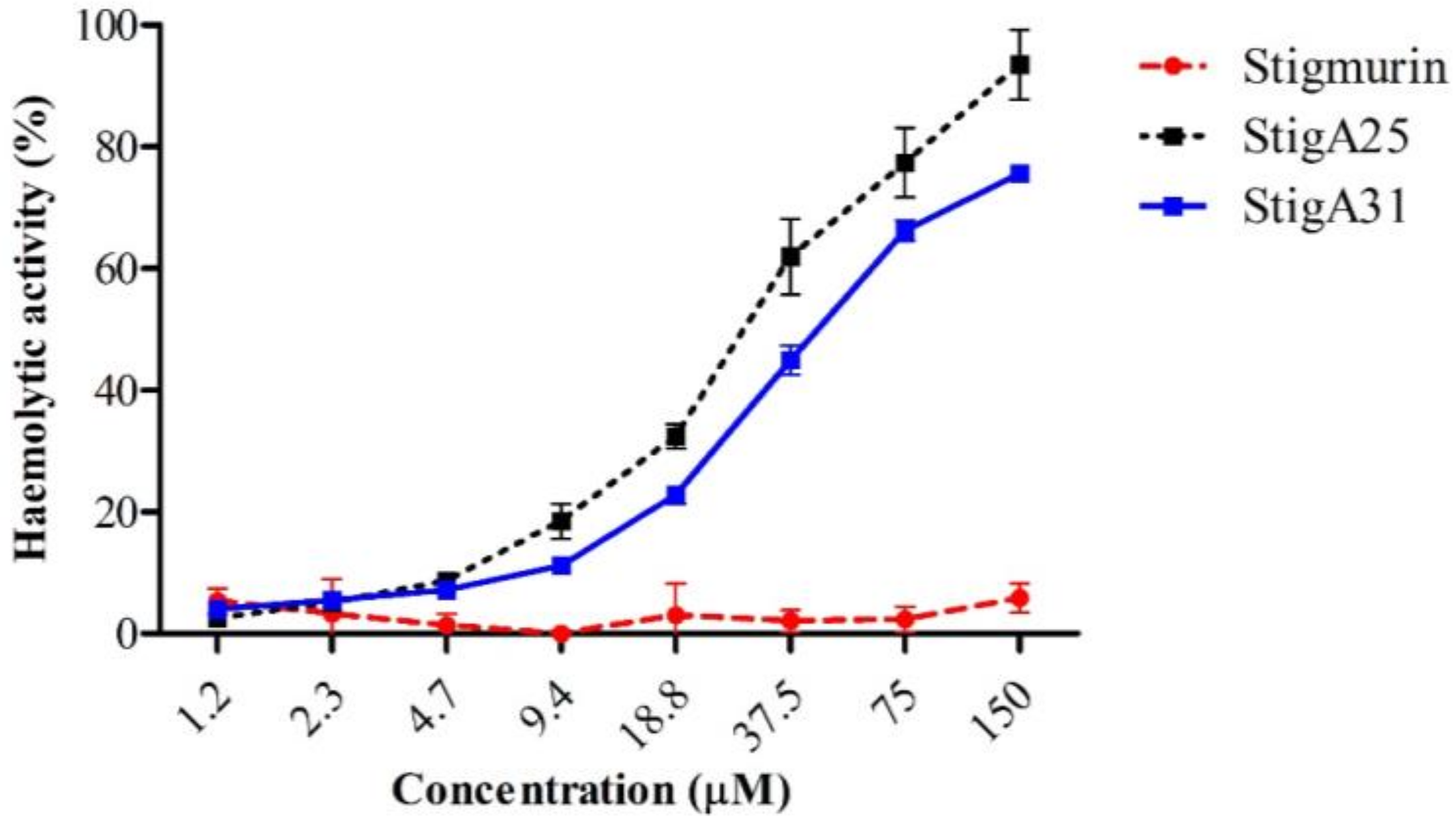
2.6. Hemolytic Activity
2.7. Antimicrobial Activity
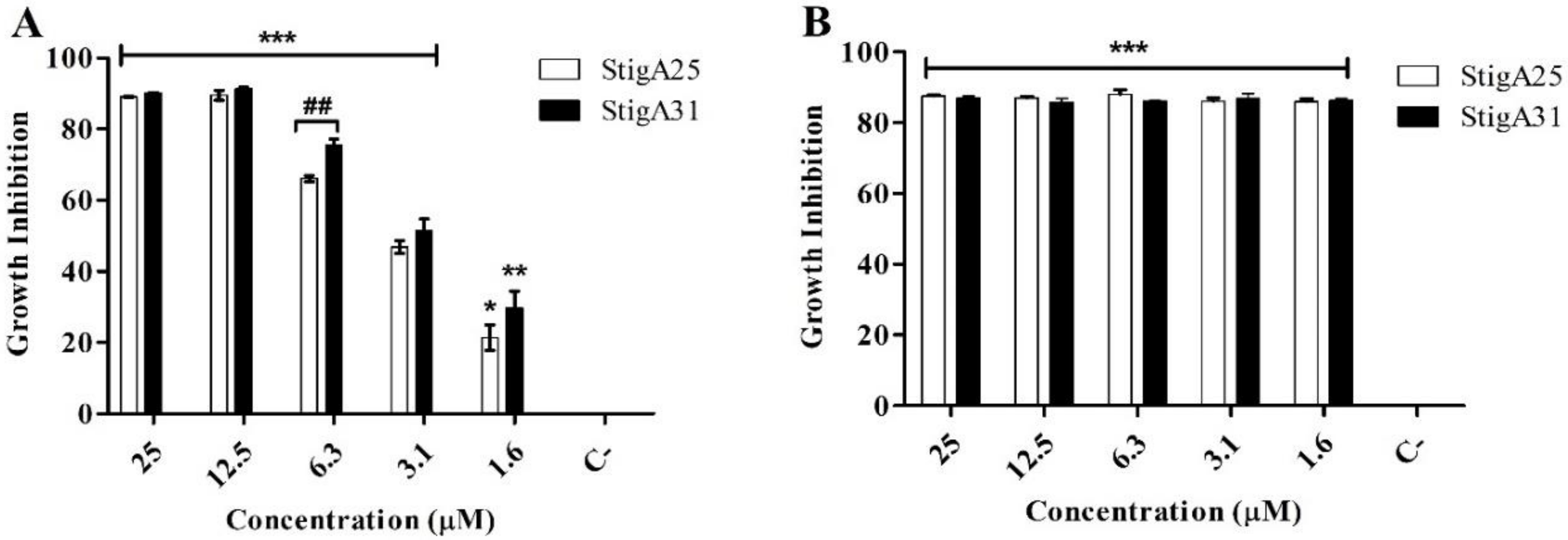
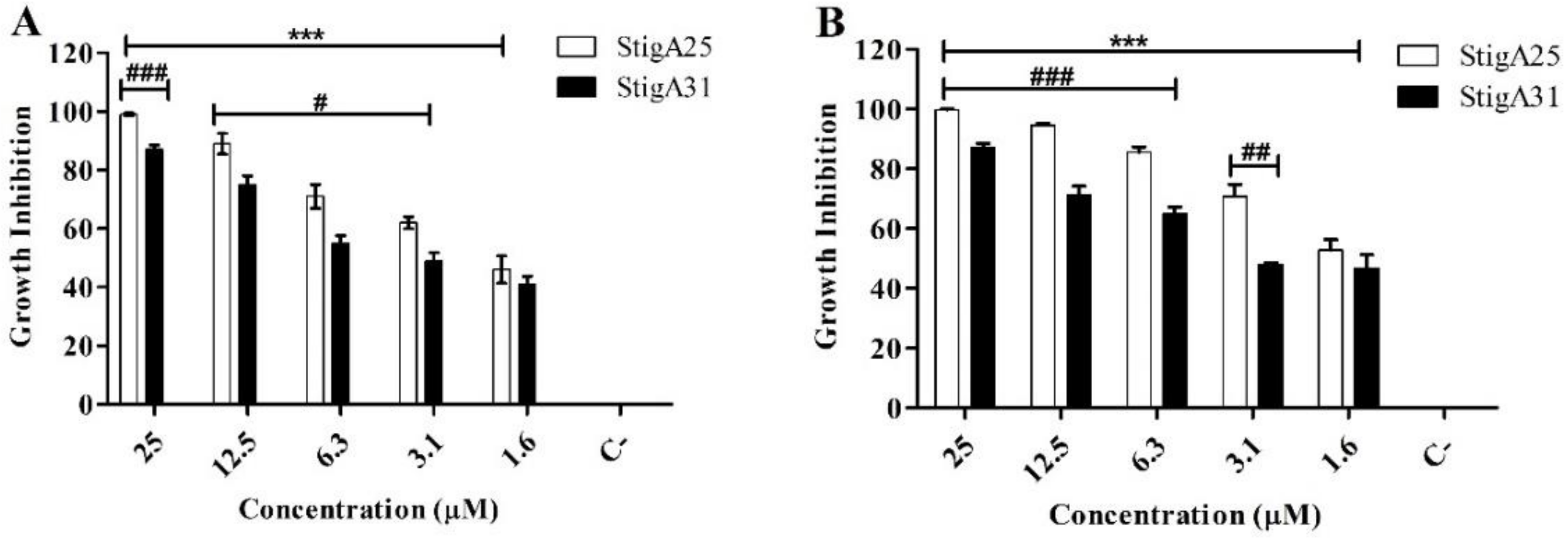
2.8. Antiparasitic Activity
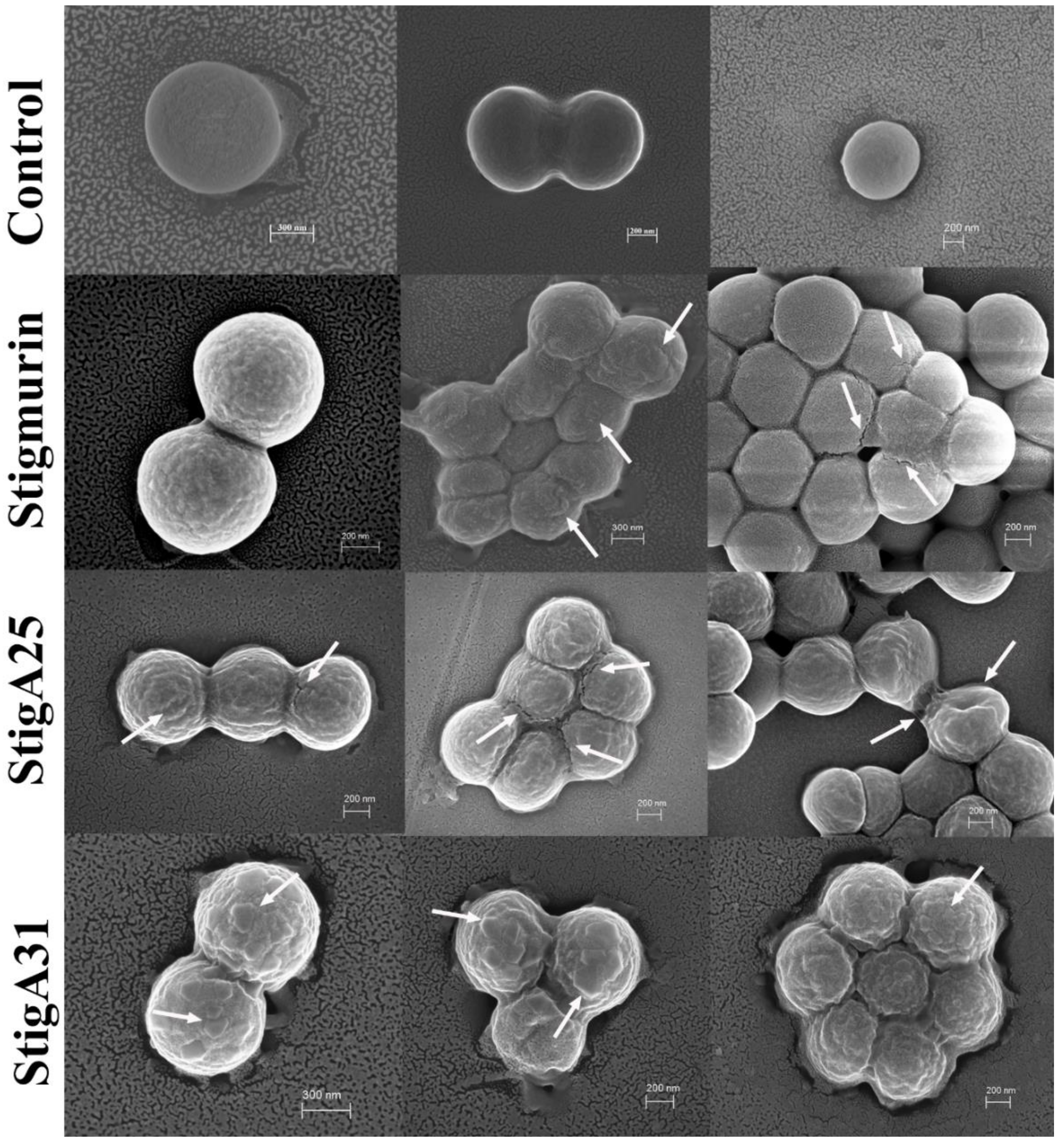
2.9. Mechanism Associated with the Antimicrobial Effect of Peptides in S. aureus
3. Discussion
4. Materials and Methods
4.1. Peptide Synthesis
4.2. In Silico Analysis
4.3. Molecular Modeling
4.4. Molecular Dynamics Simulations (MD)
4.5. Circular Dichroism Structural Characterization
4.6. Structural Stability by Circular Dichroism
4.7. Hemolytic Activity
4.8. Microorganisms
4.9. In Vitro Antimicrobial Activity
4.10. In Vitro Antiparasitic Activity
4.11. Scanning Electron Microscopy
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix







References
- Guilhelmelli, F.; Vilela, N.; Albuquerque, P.; Derengowski Lda, S.; Silva-Pereira, I.; Kyaw, C.M. Antibiotic development challenges: The various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Almeida, D.D.; Scortecci, K.C.; Kobashi, L.S.; Agnez-Lima, L.F.; Medeiros, S.R.; Silva-Junior, A.A.; Junqueira-de-Azevedo, I.; Fernandes-Pedrosa, M. Profiling the resting venom gland of the scorpion tityus stigmurus through a transcriptomic survey. BMC Genom. 2012, 13, 362. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551. [Google Scholar] [CrossRef] [PubMed]
- Mihajlovic, M.; Lazaridis, T. Charge distribution and imperfect amphipathicity affect pore formation by antimicrobial peptides. Biochim. Biophys. Acta 2012, 1818, 1274–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Johnson, W.C. Environment affects amino acid preference for secondary structure. Proc. Natl. Acad. Sci. USA 1992, 89, 4462–4465. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar] [CrossRef] [Green Version]
- Lohner, K. New strategies for novel antibiotics: Peptides targeting bacterial cell membranes. Gen. Physiol. Biophys. 2009, 28, 105–116. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. Apd2: The updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2008, 37, D933–D937. [Google Scholar] [CrossRef]
- Zhao, Z.; Ma, Y.; Dai, C.; Zhao, R.; Li, S.; Wu, Y.; Cao, Z.; Li, W. Imcroporin, a new cationic antimicrobial peptide from the venom of the scorpion isometrus maculates. Antimicrob. Agents Chemother. 2009, 53, 3472–3477. [Google Scholar] [CrossRef] [PubMed]
- Sforça, M.L.; Oyama, S.; Canduri, F.; Lorenzi, C.C.; Pertinhez, T.A.; Konno, K.; Souza, B.M.; Palma, M.S.; Ruggiero Neto, J.; Azevedo, W.F. How c-terminal carboxyamidation alters the biological activity of peptides from the venom of the eumenine solitary wasp. Biochemistry 2004, 43, 5608–5617. [Google Scholar] [CrossRef] [PubMed]
- Daniele-Silva, A.; Machado, R.J.; Monteiro, N.K.; Estrela, A.B.; Santos, E.C.; Carvalho, E.; Júnior, R.F.A.; Melo-Silveira, R.F.; Rocha, H.A.O.; Silva-Júnior, A.A. Stigmurin and tsap-2 from tityus stigmurus scorpion venom: Assessment of structure and therapeutic potential in experimental sepsis. Toxicon 2016, 121, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Melo, E.T.d.; Estrela, A.B.; Santos, E.C.G.; Machado, P.R.L.; Farias, K.J.S.; Torres, T.M.; Carvalho, E.; Lima, J.P.M.S.; Silva-Júnior, A.A.; Barbosa, E.G. Structural characterization of a novel peptide with antimicrobial activity from the venom gland of the scorpion tityus stigmurus: Stigmurin. Peptides 2015, 68, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Parente, A.; Daniele-Silva, A.; Furtado, A.A.; Melo, M.A.; Lacerda, A.F.; Queiroz, M.; Moreno, C.; Santos, E.; Rocha, H.A.; Barbosa, E.G. Analogs of the scorpion venom peptide stigmurin: Structural assessment, toxicity, and increased antimicrobial activity. Toxins 2018, 10, 161. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Kar, R.K.; Nanga, R.P.R.; Mroue, K.H.; Ramamoorthy, A.; Bhunia, A. Accelerated molecular dynamics simulation analysis of MSI-594 in a lipid bilayer. Phys. Chem. Chem. Phys. 2017, 19, 19289–19299. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef]
- Peters, B.M.; Shirtliff, M.E.; Jabra-Rizk, M.A. Antimicrobial peptides: Primeval molecules or future drugs? PLoS Pathog. 2010, 6, e1001067. [Google Scholar] [CrossRef]
- Almaaytah, A.; Albalas, Q. Scorpion venom peptides with no disulfide bridges: A review. Peptides 2014, 51, 35–45. [Google Scholar] [CrossRef]
- Santo, K.P.; Irudayam, S.J.; Berkowitz, M.L. Melittin creates transient pores in a lipid bilayer: Results from computer simulations. J. Phys. Chem. 2013, 117, 5031–5042. [Google Scholar] [CrossRef]
- Guo, X.; Ma, C.; Du, Q.; Wei, R.; Wang, L.; Zhou, M.; Chen, T.; Shaw, C. Two peptides, tsap-1 and tsap-2, from the venom of the brazilian yellow scorpion, tityus serrulatus: Evaluation of their antimicrobial and anticancer activities. Biochimie 2013, 95, 1784–1794. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, R.; Joshi, M.; Mohanram, H.; Bhunia, A.; Mangoni, M.L.; Bhattacharjya, S. Nmr structure of temporin-1 ta in lipopolysaccharide micelles: Mechanistic insight into inactivation by outer membrane. PLoS ONE 2013, 8, e72718. [Google Scholar] [CrossRef]
- Lind, T.K.; Zielinska, P.; Wacklin, H.P.; Urbanczyk-Lipkowska, Z.; Cárdenas, M. Continuous flow atomic force microscopy imaging reveals fluidity and time-dependent interactions of antimicrobial dendrimer with model lipid membranes. ACS Nano. 2013, 8, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Kotra, L.P.; Golemi, D.; Amro, N.A.; Liu, G.-Y.; Mobashery, S. Dynamics of the lipopolysaccharide assembly on the surface of escherichia c oli. J. Am. Chem. Soc. 1999, 121, 8707–8711. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Dai, C.; Li, Z.; Fan, Z.; Song, Y.; Wu, Y.; Cao, Z.; Li, W. Antibacterial activity and mechanism of a scorpion venom peptide derivative in vitro and in vivo. PLoS ONE 2012, 7, e40135. [Google Scholar] [CrossRef]
- Wu, X.; Wang, Z.; Li, X.; Fan, Y.; He, G.; Wan, Y.; Yu, C.; Tang, J.; Li, M.; Zhang, X. In vitro and in vivo activity of antimicrobial peptides developed using an amino acid-based activity prediction method. Antimicrob. Agents Chemother. 2014, 58, 5342–5349. [Google Scholar]
- Löfgren, S.; Miletti, L.; Steindel, M.; Bachere, E.; Barracco, M. Trypanocidal and leishmanicidal activities of different antimicrobial peptides (amps) isolated from aquatic animals. Exp. Parasitol. 2008, 118, 197–202. [Google Scholar] [CrossRef]
- McGwire, B.S.; Olson, C.L.; Tack, B.F.; Engman, D.M. Killing of african trypanosomes by antimicrobial peptides. J. Infect. Dis. 2003, 188, 146–152. [Google Scholar] [CrossRef]
- Zingales, B. Trypanosoma cruzi genetic diversity: Something new for something known about chagas disease manifestations, serodiagnosis and drug sensitivity. Acta Tropica. 2017. [Google Scholar] [CrossRef]
- De la Salud Bea, R.; Ascuitto, M.R.; De Johnson, L.E.L. Synthesis of analogs of peptides from buthus martensii scorpion venom with potential antibiotic activity. Peptides 2015, 68, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. Molprobity: All-atom structure validation for macromolecular crystallography. Biol. Crystal. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M. Charmm-gui membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Amos, S.-B.T.; Vermeer, L.S.; Ferguson, P.M.; Kozlowska, J.; Davy, M.; Bui, T.T.; Drake, A.F.; Lorenz, C.D.; Mason, A.J. Antimicrobial peptide potency is facilitated by greater conformational flexibility when binding to gram-negative bacterial inner membranes. Sci. Rep. 2016, 6, 37639. [Google Scholar] [CrossRef] [PubMed]
- Rončević, T.; Vukičević, D.; Ilić, N.; Krce, L.; Gajski, G.; Tonkić, M.; Goić-Barišić, I.; Zoranić, L.; Sonavane, Y.; Benincasa, M. Antibacterial activity affected by the conformational flexibility in glycine–lysine based α-helical antimicrobial peptides. J. Med. Chem. 2018, 61, 2924–2936. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D. Charmm36 all-atom additive protein force field: Validation based on comparison to nmr data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. The Journal of chemical physics 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Berendsen, H. A leap-frog algorithm for stochastic dynamics. Mol. Simulation 1988, 1, 173–185. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald: An n⋅ log (n) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. Lincs: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Physical Rev. 1985, 31, 1695. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Ribeiro, L.F.; Tullman, J.; Nicholes, N.; Silva, S.R.B.; Vieira, D.S.; Ostermeier, M.; Ward, R.J. A xylose-stimulated xylanase–xylose binding protein chimera created by random nonhomologous recombination. Biotechnol. Biofuels 2016, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.B.; Pinheiro, M.P.; Fuzo, C.A.; Silva, S.R.; Ferreira, T.L.; Lourenzoni, M.R.; Nonato, M.C.; Vieira, D.S.; Ward, R.J. The role of local residue environmental changes in thermostable mutants of the gh11 xylanase from bacillus subtilis. Int. J. Biol. Macromol. 2017, 97, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Tzen, J.T.; Shyu, C.-L.; Yang, M.J.; Tu, W.-C. Structural and biological characterization of mastoparans in the venom of vespa species in taiwan. Peptides 2011, 32, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of contin, selcon, and cdsstr methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef]
- Menezes, Y.A.; Félix-Silva, J.; Da Silva-Júnior, A.A.; Rebecchi, I.M.; De Oliveira, A.S.; Uchoa, A.F.; Fernandes-Pedrosa, M.d.F. Protein-rich fraction of cnidoscolus urens (l.) arthur leaves: Enzymatic characterization and procoagulant and fibrinogenolytic activities. Molecules 2014, 19, 3552–3569. [Google Scholar] [CrossRef]
- Cockerill, F.R. Performance Standards for Antimicrobial Susceptibility Testing: Twenty-First Informational Supplement; Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2011. [Google Scholar]
- Cos, P.; Vlietinck, A.J.; Berghe, D.V.; Maes, L. Anti-infective potential of natural products: How to develop a stronger in vitro ‘proof-of-concept’. J. Ethnopharmacol. 2006, 106, 290–302. [Google Scholar] [CrossRef]
- Flores-Solis, D.; Toledano, Y.; Rodríguez-Lima, O.; Cano-Sánchez, P.; Ramírez-Cordero, B.E.; Landa, A.; Rodríguez de la Vega, R.C.; Rio-Portilla, F. Solution structure and antiparasitic activity of scorpine-like peptides from hoffmannihadrurus gertschi. FEBS Lett. 2016, 590, 2286–2296. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
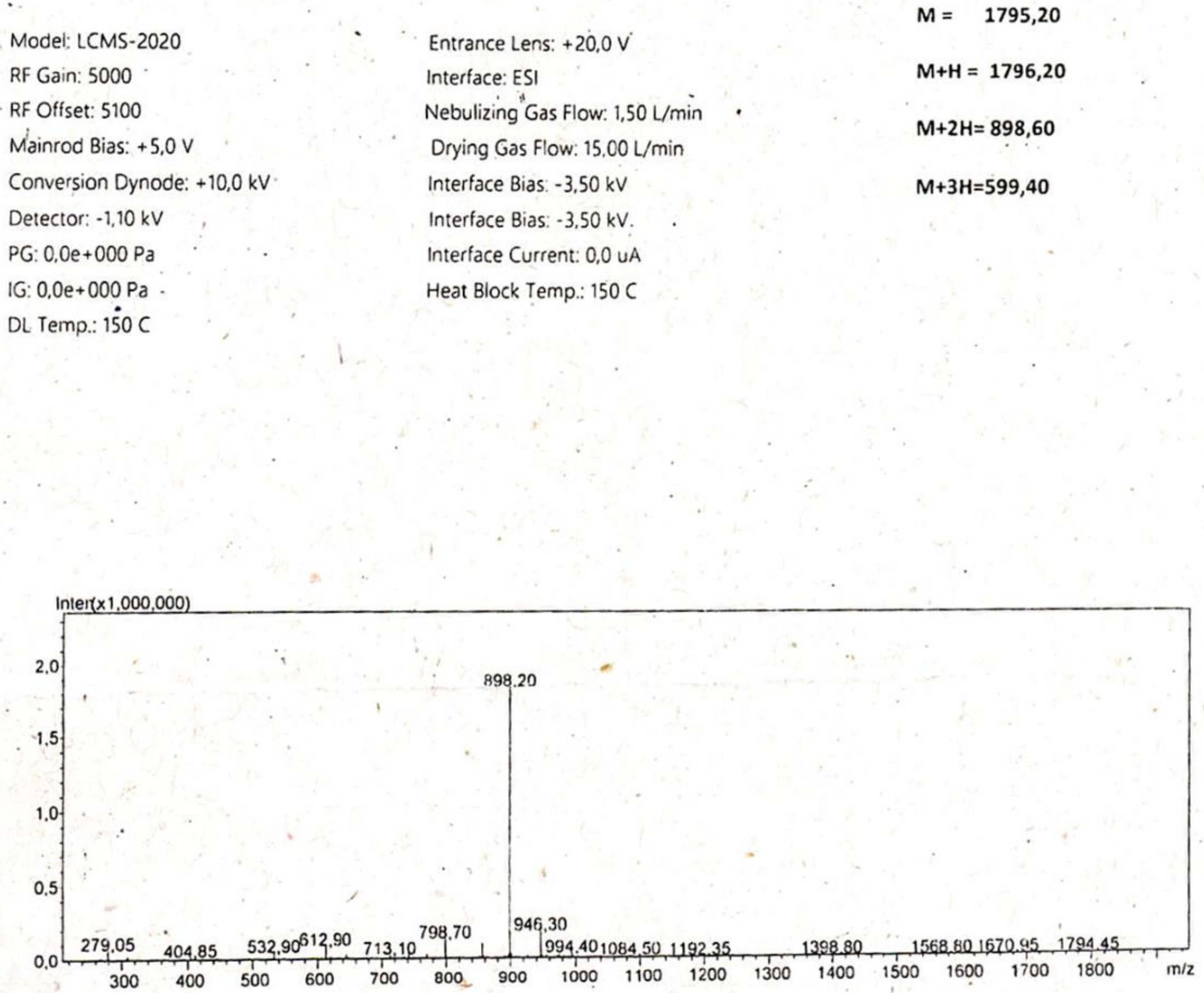{kind=link}
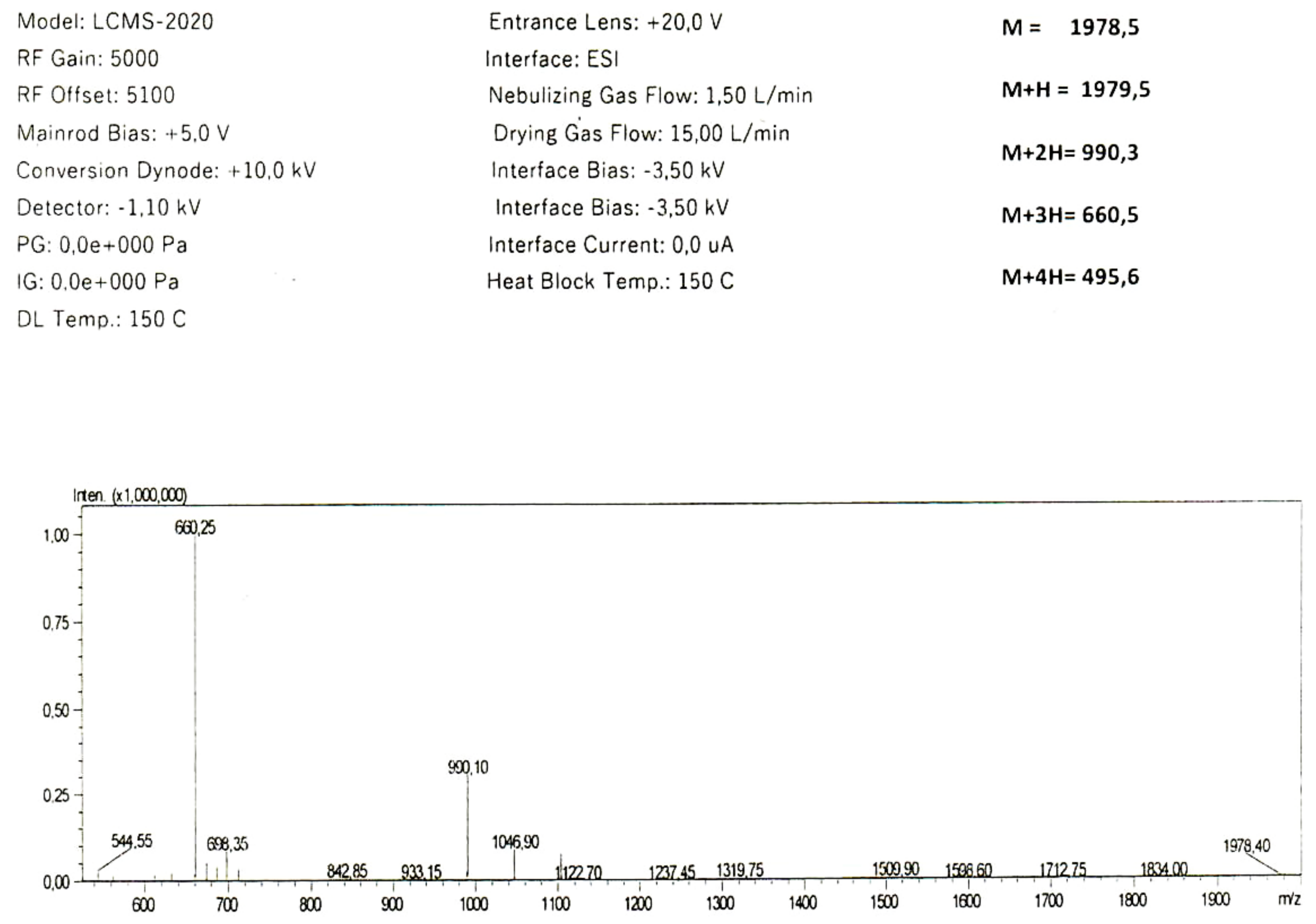{kind=link}
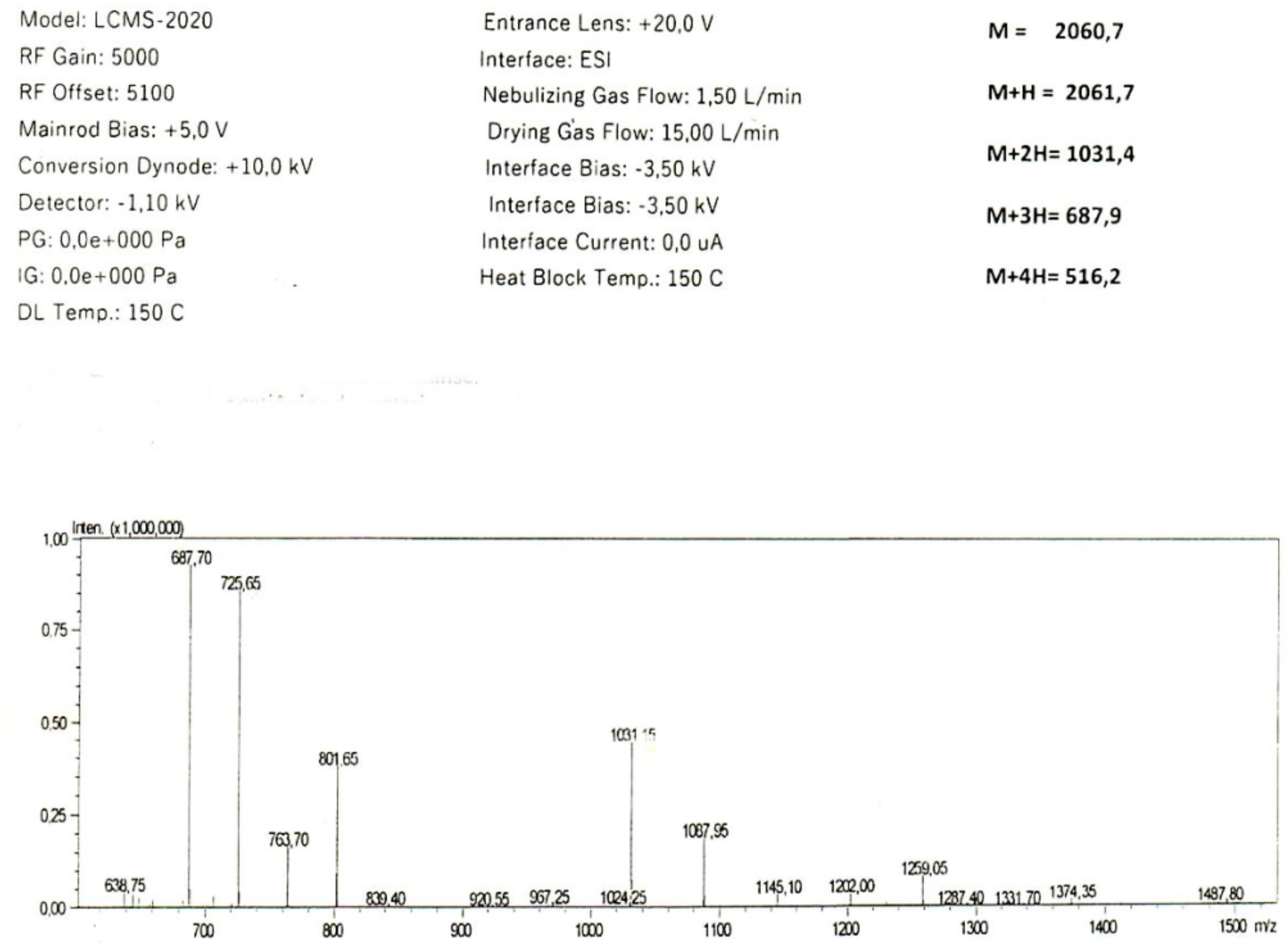{kind=link}
{kind=link}
| Peptides | Sequences a | Secondary Structure b | α-Helix | H | µH | Charge |
|---|---|---|---|---|---|---|
| Stigmurin | FFSLIPSLVGGLISAFK-NH2 | CCCCCHHHHHHHHHHHC | 64.7 % | 0.89 | 0.57 | +2 |
| StigA25 | FFSLIPSLVKKLIKAFK-NH2 | CCCHHHHHHHHHHHCC | 70.5 % | 0.73 | 0.70 | +5 |
| StigA31 | FFKLIPKLVKKLIKAFK-NH2 | CHHHHHHHHHHHHHHHC | 88.2 % | 0.61 | 0.80 | +7 |
| Solvent | α-Helical % | β-Sheet % | Random Coil % | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Stig * | StigA25 | StigA31 | Stig * | StigA25 | StigA31 | Stig * | StigA25 | StigA31 | |
| Water | 3 | 2 | 2 | 17 | 25 | 17 | 79 | 72 | 79 |
| PBS 10 mM | 3 | 2 | 2 | 13 | 28 | 19 | 82 | 68 | 78 |
| SDS 20 mM | 68 | 41 | 54 | 4 | 18 | 9 | 27 | 41 | 36 |
| SDS 4 mM ** | 65 | 42 | 56 | 6 | 19 | 15 | 28 | 40 | 28 |
| TFE 20% | 58 | 40 | 49 | 10 | 17 | 16 | 32 | 42 | 34 |
| TFE 30% | 68 | 41 | 52 | 7 | 19 | 14 | 24 | 40 | 35 |
| TFE 40% | 70 | 40 | 49 | 6 | 18 | 17 | 23 | 41 | 33 |
| TFE 50% | 65 | 41 | 50 | 6 | 21 | 15 | 28 | 39 | 33 |
| TFE 60% | 59 | 43 | 49 | 12 | 17 | 15 | 29 | 40 | 35 |
| TFE 70% | 59 | 39 | 46 | 11 | 19 | 16 | 29 | 41 | 17 |
| Potential of Hydrogen (pH) | α-Helical % | β-Sheet % | Random Coil % | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Stig * | StigA25 | StigA31 | Stig * | StigA25 | StigA31 | Stig * | StigA25 | StigA31 | |
| 3 | 70 | 50 | 53 | 5 | 12 | 11 | 24 | 37 | 36 |
| 4 | 69 | 49 | 54 | 6 | 13 | 12 | 25 | 37 | 33 |
| 5 | 69 | 58 | 59 | 5 | 16 | 12 | 26 | 27 | 29 |
| 6 | 76 | 48 | 52 | 10 | 14 | 13 | 15 | 39 | 35 |
| 7 | 69 | 49 | 54 | 5 | 14 | 13 | 26 | 36 | 34 |
| 7.4 | 79 | 47 | 53 | 5 | 16 | 12 | 16 | 36 | 35 |
| 8 | 69 | 49 | 51 | 6 | 13 | 12 | 24 | 37 | 36 |
| 9 | 80 | 51 | 57 | 5 | 12 | 12 | 14 | 36 | 30 |
| Strains | Minimum Inhibitory Concentration (MIC) | |||
|---|---|---|---|---|
| Stigmurin (µM) | StigA25 (µM) | StigA31 (µM) | Antibiotics (µM) * | |
| Gram-positive | ||||
| Staphylococcus aureus (ATCC 29213) | 9.4 | 1.2 | 2.3 | 5.5 |
| Staphylococcus epidermidis (ATCC 12228) | 9.4 | 2.3 | 2.3 | 22.1 |
| Enterococcus faecalis (ATCC 4028) | >150 | 4.7 | 1.2 | 5.5 |
| Gram-negative | ||||
| Pseudomonas aeruginosa (ATCC 27853) | >150 | 4.7 | 2.3 | 8.4 |
| Escherichia coli (ATCC 25922) | >150 | 2.3 | 1.2 | 4.2 |
| Enterobacter cloacae (ATCC 13047) | >150 | 18.8 | 4.7 | 67 |
| Yeasts | ||||
| Candida albicans (ATCC 90028) | 37.5 | 9.4 | 4.7 | 34 |
| Candida glabrata (ATCC 90030) | >150 | 9.4 | 4.7 | 3.2 |
| Candida krusei (ATCC 6258) | >150 | 9.4 | 4.7 | 34 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amorim-Carmo, B.; Daniele-Silva, A.; Parente, A.M.S.; Furtado, A.A.; Carvalho, E.; Oliveira, J.W.F.; Santos, E.C.G.; Silva, M.S.; Silva, S.R.B.; Silva-Júnior, A.A.; et al. Potent and Broad-Spectrum Antimicrobial Activity of Analogs from the Scorpion Peptide Stigmurin. Int. J. Mol. Sci. 2019, 20, 623. https://doi.org/10.3390/ijms20030623
Amorim-Carmo B, Daniele-Silva A, Parente AMS, Furtado AA, Carvalho E, Oliveira JWF, Santos ECG, Silva MS, Silva SRB, Silva-Júnior AA, et al. Potent and Broad-Spectrum Antimicrobial Activity of Analogs from the Scorpion Peptide Stigmurin. International Journal of Molecular Sciences. 2019; 20(3):623. https://doi.org/10.3390/ijms20030623
Chicago/Turabian StyleAmorim-Carmo, Bruno, Alessandra Daniele-Silva, Adriana M. S. Parente, Allanny A. Furtado, Eneas Carvalho, Johny W. F. Oliveira, Elizabeth C. G. Santos, Marcelo S. Silva, Sérgio R. B. Silva, Arnóbio A. Silva-Júnior, and et al. 2019. "Potent and Broad-Spectrum Antimicrobial Activity of Analogs from the Scorpion Peptide Stigmurin" International Journal of Molecular Sciences 20, no. 3: 623. https://doi.org/10.3390/ijms20030623
APA StyleAmorim-Carmo, B., Daniele-Silva, A., Parente, A. M. S., Furtado, A. A., Carvalho, E., Oliveira, J. W. F., Santos, E. C. G., Silva, M. S., Silva, S. R. B., Silva-Júnior, A. A., Monteiro, N. K., & Fernandes-Pedrosa, M. F. (2019). Potent and Broad-Spectrum Antimicrobial Activity of Analogs from the Scorpion Peptide Stigmurin. International Journal of Molecular Sciences, 20(3), 623. https://doi.org/10.3390/ijms20030623