A Novel Insight on Endotyping Heterogeneous Severe Asthma Based on Endoplasmic Reticulum Stress: Beyond the “Type 2/Non-Type 2 Dichotomy”


{kind=link}
{kind=link}
Abstract
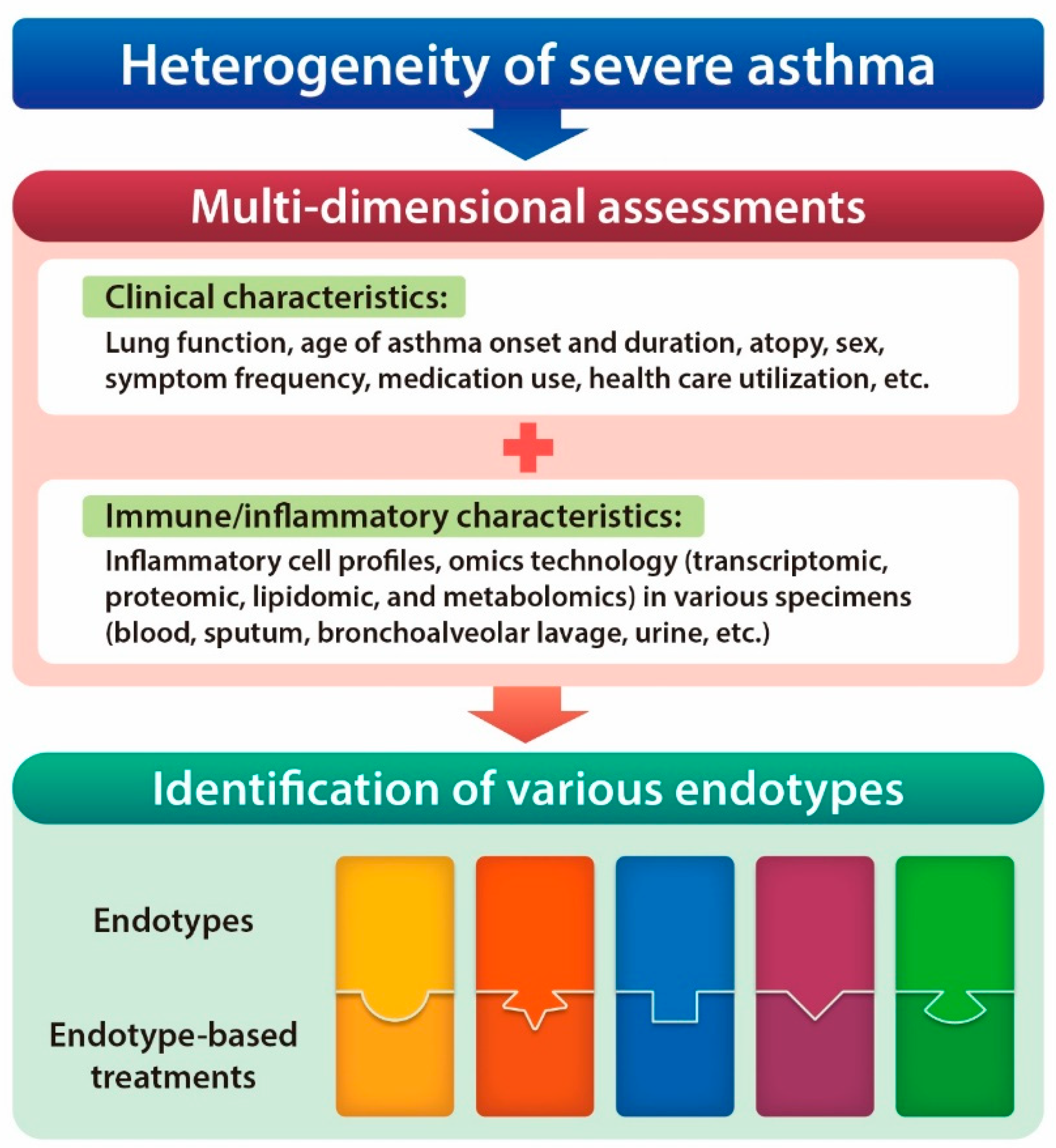
:1. Introduction
2. Type 2 and Non-Type 2 Immune Responses in the Heterogeneity of Severe Asthma
3. Type 2 Inflammation: Allergic and Non-Allergic Eosinophilic Airway Inflammation
4. Non-Type 2 Inflammation: Neutrophilic Airway Inflammation in Association with Type 2 Immune Response
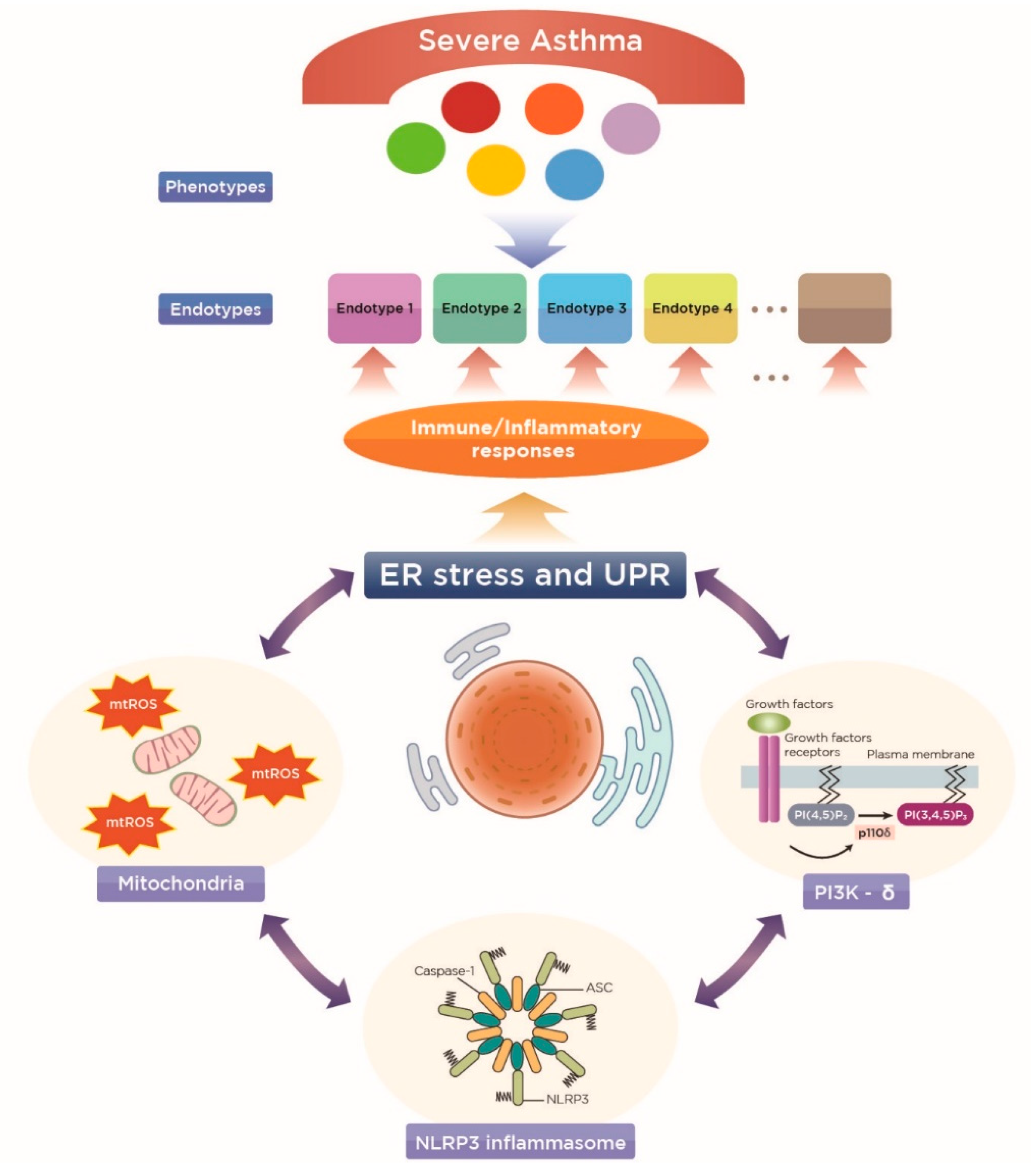
4.1. A New Perspective on Endotyping Heterogeneous Severe Asthma: Implication of Subcellular Organelles
4.2. ER Stress as a Potential Endotype of Severe Asthma
5. Implications of Mitochondria and the Related Cellular Immune/Inflammatory Platforms in ER-Associated Endotypes of Severe Asthma
5.1. Mitochondria
5.2. NLRP3 Inflammasome
5.3. Phosphoinositide 3-Kinase-δ (PI3K-δ)
6. Lessons Learned from Sputum Transcriptomic Data of U-BIOPRED
7. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Agache, I.; Akdis, C.A. Endotypes of allergic diseases and asthma: An important step in building blocks for the future of precision medicine. Allergol. Int. 2016, 65, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Fajt, M.L.; Wenzel, S.E. Asthma phenotypes and the use of biologic medications in asthma and allergic disease: The next steps toward personalized care. J. Allergy Clin. Immunol. 2015, 135, 299–310; quiz 311. [Google Scholar] [CrossRef]
- Kupczyk, M.; Wenzel, S. U.S. and European severe asthma cohorts: What can they teach us about severe asthma? J. Intern. Med. 2012, 272, 121–132. [Google Scholar] [CrossRef]
- Lang, D.M. Severe asthma: Epidemiology, burden of illness, and heterogeneity. Allergy Asthma Proc. 2015, 36, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Moore, W.C.; Meyers, D.A.; Wenzel, S.E.; Teague, W.G.; Li, H.; Li, X.; D’Agostino, R., Jr.; Castro, M.; Curran-Everett, D.; Fitzpatrick, A.M.; et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am. J. Respir. Crit. Care Med. 2010, 181, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Hastie, A.T.; Moore, W.C.; Meyers, D.A.; Vestal, P.L.; Li, H.; Peters, S.P.; Bleecker, E.R. Analyses of asthma severity phenotypes and inflammatory proteins in subjects stratified by sputum granulocytes. J. Allergy Clin. Immunol. 2010, 125, 1028–1036.e1013. [Google Scholar] [CrossRef] [PubMed]
- Moore, W.C.; Fitzpatrick, A.M.; Li, X.; Hastie, A.T.; Li, H.; Meyers, D.A.; Bleecker, E.R. Clinical heterogeneity in the severe asthma research program. Ann. Am. Thorac. Soc. 2013, 10, S118–S124. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The immunology of asthma. Nat. Immunol. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Carr, T.F.; Zeki, A.A.; Kraft, M. Eosinophilic and Noneosinophilic Asthma. Am. J. Respir. Crit. Care Med. 2018, 197, 22–37. [Google Scholar] [CrossRef]
- Wu, W.; Bleecker, E.; Moore, W.; Busse, W.W.; Castro, M.; Chung, K.F.; Calhoun, W.J.; Erzurum, S.; Gaston, B.; Israel, E.; et al. Unsupervised phenotyping of Severe Asthma Research Program participants using expanded lung data. J. Allergy Clin. Immunol. 2014, 133, 1280–1288. [Google Scholar] [CrossRef]
- Kuo, C.S.; Pavlidis, S.; Loza, M.; Baribaud, F.; Rowe, A.; Pandis, I.; Sousa, A.; Corfield, J.; Djukanovic, R.; Lutter, R.; et al. T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur. Respir. J. 2017, 49, 1602135. [Google Scholar] [CrossRef]
- Jeong, J.S.; Kim, S.R.; Lee, Y.C. Can Controlling Endoplasmic Reticulum Dysfunction Treat Allergic Inflammation in Severe Asthma with Fungal Sensitization? Allergy Asthma Immunol. Res. 2018, 10, 106–120. [Google Scholar] [CrossRef]
- Jeong, J.S.; Kim, S.R.; Cho, S.H.; Lee, Y.C. Endoplasmic Reticulum Stress and Allergic Diseases. Curr. Allergy Asthma Rep. 2017, 17, 82. [Google Scholar] [CrossRef]
- Muraro, A.; Lemanske, R.F., Jr.; Hellings, P.W.; Akdis, C.A.; Bieber, T.; Casale, T.B.; Jutel, M.; Ong, P.Y.; Poulsen, L.K.; Schmid-Grendelmeier, P.; et al. Precision medicine in patients with allergic diseases: Airway diseases and atopic dermatitis-PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J. Allergy Clin. Immunol. 2016, 137, 1347–1358. [Google Scholar]
- Grunig, G.; Warnock, M.; Wakil, A.E.; Venkayya, R.; Brombacher, F.; Rennick, D.M.; Sheppard, D.; Mohrs, M.; Donaldson, D.D.; Locksley, R.M.; et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science 1998, 282, 2261–2263. [Google Scholar] [CrossRef]
- Chu, D.K.; Jimenez-Saiz, R.; Verschoor, C.P.; Walker, T.D.; Goncharova, S.; Llop-Guevara, A.; Shen, P.; Gordon, M.E.; Barra, N.G.; Bassett, J.D.; et al. Indigenous enteric eosinophils control DCs to initiate a primary Th2 immune response in vivo. J. Exp. Med. 2014, 211, 1657–1672. [Google Scholar] [CrossRef]
- Shi, H.Z.; Humbles, A.; Gerard, C.; Jin, Z.; Weller, P.F. Lymph node trafficking and antigen presentation by endobronchial eosinophils. J. Clin. Investig. 2000, 105, 945–953. [Google Scholar] [CrossRef]
- Coyle, A.J.; Ackerman, S.J.; Burch, R.; Proud, D.; Irvin, C.G. Human eosinophil-granule major basic protein and synthetic polycations induce airway hyperresponsiveness in vivo dependent on bradykinin generation. J. Clin. Investig. 1995, 95, 1735–1740. [Google Scholar] [CrossRef]
- Song, D.J.; Cho, J.Y.; Lee, S.Y.; Miller, M.; Rosenthal, P.; Soroosh, P.; Croft, M.; Zhang, M.; Varki, A.; Broide, D.H. Anti-Siglec-F antibody reduces allergen-induced eosinophilic inflammation and airway remodeling. J. Immunol. 2009, 183, 5333–5341. [Google Scholar] [CrossRef]
- Dweik, R.A.; Boggs, P.B.; Erzurum, S.C.; Irvin, C.G.; Leigh, M.W.; Lundberg, J.O.; Olin, A.C.; Plummer, A.L.; Taylor, D.R.; American Thoracic Society Committee on Interpretation of Exhaled Nitric Oxide Levels (FENO) for Clinical Applications. An official ATS clinical practice guideline: Interpretation of exhaled nitric oxide levels (FENO) for clinical applications. Am. J. Respir. Crit. Care Med. 2011, 184, 602–615. [Google Scholar] [CrossRef]
- Cheng, D.; Xue, Z.; Yi, L.; Shi, H.; Zhang, K.; Huo, X.; Bonser, L.R.; Zhao, J.; Xu, Y.; Erle, D.J.; et al. Epithelial interleukin-25 is a key mediator in Th2-high, corticosteroid-responsive asthma. Am. J. Respir. Crit. Care Med. 2014, 190, 639–648. [Google Scholar] [CrossRef]
- Jia, G.; Erickson, R.W.; Choy, D.F.; Mosesova, S.; Wu, L.C.; Solberg, O.D.; Shikotra, A.; Carter, R.; Audusseau, S.; Hamid, Q.; et al. Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J. Allergy Clin. Immunol. 2012, 130, 647–654.e610. [Google Scholar] [CrossRef]
- Doherty, T.A.; Khorram, N.; Lund, S.; Mehta, A.K.; Croft, M.; Broide, D.H. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J. Allergy Clin. Immunol. 2013, 132, 205–213. [Google Scholar] [CrossRef]
- Xue, L.; Salimi, M.; Panse, I.; Mjosberg, J.M.; McKenzie, A.N.; Spits, H.; Klenerman, P.; Ogg, G. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J. Allergy Clin. Immunol. 2014, 133, 1184–1194. [Google Scholar] [CrossRef]
- Kabata, H.; Moro, K.; Fukunaga, K.; Suzuki, Y.; Miyata, J.; Masaki, K.; Betsuyaku, T.; Koyasu, S.; Asano, K. Thymic stromal lymphopoietin induces corticosteroid resistance in natural helper cells during airway inflammation. Nat. Commun. 2013, 4, 2675. [Google Scholar] [CrossRef]
- Liu, S.; Verma, M.; Michalec, L.; Liu, W.; Sripada, A.; Rollins, D.; Good, J.; Ito, Y.; Chu, H.; Gorska, M.M.; et al. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: The role of thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 2018, 141, 257–268.e256. [Google Scholar] [CrossRef]
- Halim, T.Y.; Steer, C.A.; Matha, L.; Gold, M.J.; Martinez-Gonzalez, I.; McNagny, K.M.; McKenzie, A.N.; Takei, F. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 2014, 40, 425–435. [Google Scholar] [CrossRef]
- Oliphant, C.J.; Hwang, Y.Y.; Walker, J.A.; Salimi, M.; Wong, S.H.; Brewer, J.M.; Englezakis, A.; Barlow, J.L.; Hams, E.; Scanlon, S.T.; et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 2014, 41, 283–295. [Google Scholar] [CrossRef]
- Ortega, H.G.; Liu, M.C.; Pavord, I.D.; Brusselle, G.G.; FitzGerald, J.M.; Chetta, A.; Humbert, M.; Katz, L.E.; Keene, O.N.; Yancey, S.W.; et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N. Engl. J. Med. 2014, 371, 1198–1207. [Google Scholar] [CrossRef]
- Castro, M.; Zangrilli, J.; Wechsler, M.E.; Bateman, E.D.; Brusselle, G.G.; Bardin, P.; Murphy, K.; Maspero, J.F.; O’Brien, C.; Korn, S. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: Results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir. Med. 2015, 3, 355–366. [Google Scholar] [CrossRef]
- FitzGerald, J.M.; Bleecker, E.R.; Nair, P.; Korn, S.; Ohta, K.; Lommatzsch, M.; Ferguson, G.T.; Busse, W.W.; Barker, P.; Sproule, S.; et al. Benralizumab, an anti-interleukin-5 receptor alpha monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2016, 388, 2128–2141. [Google Scholar] [CrossRef]
- Woodruff, P.G.; Modrek, B.; Choy, D.F.; Jia, G.; Abbas, A.R.; Ellwanger, A.; Koth, L.L.; Arron, J.R.; Fahy, J.V. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am. J. Respir. Crit. Care Med. 2009, 180, 388–395. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Schwartz, L.B.; Langmack, E.L.; Halliday, J.L.; Trudeau, J.B.; Gibbs, R.L.; Chu, H.W. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am. J. Respir. Crit. Care Med. 1999, 160, 1001–1008. [Google Scholar] [CrossRef]
- Samitas, K.; Zervas, E.; Gaga, M. T2-low asthma: Current approach to diagnosis and therapy. Curr. Opin. Pulm. Med. 2017, 23, 48–55. [Google Scholar] [CrossRef]
- Moore, W.C.; Hastie, A.T.; Li, X.; Li, H.; Busse, W.W.; Jarjour, N.N.; Wenzel, S.E.; Peters, S.P.; Meyers, D.A.; Bleecker, E.R. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J. Allergy Clin. Immunol. 2014, 133, 1557–1563.e1555. [Google Scholar] [CrossRef]
- Corren, J.; Busse, W.; Meltzer, E.O.; Mansfield, L.; Bensch, G.; Fahrenholz, J.; Wenzel, S.E.; Chon, Y.; Dunn, M.; Weng, H.H.; et al. A randomized, controlled, phase 2 study of AMG 317, an IL-4Ralpha antagonist, in patients with asthma. Am. J. Respir. Crit. Care Med. 2010, 181, 788–796. [Google Scholar] [CrossRef]
- Al-Ramli, W.; Prefontaine, D.; Chouiali, F.; Martin, J.G.; Olivenstein, R.; Lemiere, C.; Hamid, Q. T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J. Allergy Clin. Immunol. 2009, 123, 1185–1187. [Google Scholar] [CrossRef]
- McKinley, L.; Alcorn, J.F.; Peterson, A.; Dupont, R.B.; Kapadia, S.; Logar, A.; Henry, A.; Irvin, C.G.; Piganelli, J.D.; Ray, A.; et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 2008, 181, 4089–4097. [Google Scholar] [CrossRef]
- Shaw, D.E.; Berry, M.A.; Hargadon, B.; McKenna, S.; Shelley, M.J.; Green, R.H.; Brightling, C.E.; Wardlaw, A.J.; Pavord, I.D. Association between neutrophilic airway inflammation and airflow limitation in adults with asthma. Chest 2007, 132, 1871–1875. [Google Scholar] [CrossRef]
- Raundhal, M.; Morse, C.; Khare, A.; Oriss, T.B.; Milosevic, J.; Trudeau, J.; Huff, R.; Pilewski, J.; Holguin, F.; Kolls, J.; et al. High IFN-gamma and low SLPI mark severe asthma in mice and humans. J. Clin. Investig. 2015, 125, 3037–3050. [Google Scholar] [CrossRef]
- Chambers, E.S.; Nanzer, A.M.; Pfeffer, P.E.; Richards, D.F.; Timms, P.M.; Martineau, A.R.; Griffiths, C.J.; Corrigan, C.J.; Hawrylowicz, C.M. Distinct endotypes of steroid-resistant asthma characterized by IL-17A(high) and IFN-gamma(high) immunophenotypes: Potential benefits of calcitriol. J. Allergy Clin. Immunol. 2015, 136, 628–637.e624. [Google Scholar] [CrossRef]
- Duvall, M.G.; Barnig, C.; Cernadas, M.; Ricklefs, I.; Krishnamoorthy, N.; Grossman, N.L.; Bhakta, N.R.; Fahy, J.V.; Bleecker, E.R.; Castro, M.; et al. Natural killer cell-mediated inflammation resolution is disabled in severe asthma. Sci. Immunol. 2017, 2, eaam5446. [Google Scholar] [CrossRef]
- Randolph, D.A.; Stephens, R.; Carruthers, C.J.; Chaplin, D.D. Cooperation between Th1 and Th2 cells in a murine model of eosinophilic airway inflammation. J. Clin. Investig. 1999, 104, 1021–1029. [Google Scholar] [CrossRef]
- Ford, J.G.; Rennick, D.; Donaldson, D.D.; Venkayya, R.; McArthur, C.; Hansell, E.; Kurup, V.P.; Warnock, M.; Grunig, G. Il-13 and IFN-gamma: Interactions in lung inflammation. J. Immunol. 2001, 167, 1769–1777. [Google Scholar] [CrossRef]
- Park, S.J.; Lee, K.S.; Kim, S.R.; Min, K.H.; Choe, Y.H.; Moon, H.; Chae, H.J.; Yoo, W.H.; Lee, Y.C. Peroxisome proliferator-activated receptor gamma agonist down-regulates IL-17 expression in a murine model of allergic airway inflammation. J. Immunol. 2009, 183, 3259–3267. [Google Scholar] [CrossRef]
- Park, S.J.; Lee, K.S.; Kim, S.R.; Min, K.H.; Moon, H.; Lee, M.H.; Chung, C.R.; Han, H.J.; Puri, K.D.; Lee, Y.C. Phosphoinositide 3-kinase delta inhibitor suppresses interleukin-17 expression in a murine asthma model. Eur. Respir. J. 2010, 36, 1448–1459. [Google Scholar] [CrossRef]
- Ray, A.; Kolls, J.K. Neutrophilic Inflammation in Asthma and Association with Disease Severity. Trends Immunol. 2017, 38, 942–954. [Google Scholar] [CrossRef]
- Ray, A.; Raundhal, M.; Oriss, T.B.; Ray, P.; Wenzel, S.E. Current concepts of severe asthma. J. Clin. Investig. 2016, 126, 2394–2403. [Google Scholar] [CrossRef]
- Busse, W.W.; Holgate, S.; Kerwin, E.; Chon, Y.; Feng, J.; Lin, J.; Lin, S.L. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am. J. Respir. Crit. Care Med. 2013, 188, 1294–1302. [Google Scholar] [CrossRef]
- Pathinayake, P.S.; Hsu, A.C.; Waters, D.W.; Hansbro, P.M.; Wood, L.G.; Wark, P.A.B. Understanding the Unfolded Protein Response in the Pathogenesis of Asthma. Front. Immunol. 2018, 9, 175. [Google Scholar] [CrossRef]
- Vannuvel, K.; Renard, P.; Raes, M.; Arnould, T. Functional and morphological impact of ER stress on mitochondria. J. Cell. Physiol. 2013, 228, 1802–1818. [Google Scholar] [CrossRef]
- Osorio, F.; Lambrecht, B.; Janssens, S. The UPR and lung disease. Semin. Immunopathol. 2013, 35, 293–306. [Google Scholar] [CrossRef]
- Bettigole, S.E.; Glimcher, L.H. Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 2015, 33, 107–138. [Google Scholar] [CrossRef]
- Kim, S.R.; Kim, D.I.; Kang, M.R.; Lee, K.S.; Park, S.Y.; Jeong, J.S.; Lee, Y.C. Endoplasmic reticulum stress influences bronchial asthma pathogenesis by modulating nuclear factor kappaB activation. J. Allergy Clin. Immunol. 2013, 132, 1397–1408. [Google Scholar] [CrossRef]
- Guo, Q.; Li, H.; Liu, J.; Xu, L.; Yang, L.; Sun, Z.; Zhou, B. Tunicamycin aggravates endoplasmic reticulum stress and airway inflammation via PERK-ATF4-CHOP signaling in a murine model of neutrophilic asthma. J. Asthma 2017, 54, 125–133. [Google Scholar] [CrossRef]
- Lee, K.S.; Jeong, J.S.; Kim, S.R.; Cho, S.H.; Kolliputi, N.; Ko, Y.H.; Lee, K.B.; Park, S.C.; Park, H.J.; Lee, Y.C. Phosphoinositide 3-kinase-delta regulates fungus-induced allergic lung inflammation through endoplasmic reticulum stress. Thorax 2016, 71, 52–63. [Google Scholar] [CrossRef]
- Lotvall, J.; Akdis, C.A.; Bacharier, L.B.; Bjermer, L.; Casale, T.B.; Custovic, A.; Lemanske, R.F., Jr.; Wardlaw, A.J.; Wenzel, S.E.; Greenberger, P.A. Asthma endotypes: A new approach to classification of disease entities within the asthma syndrome. J. Allergy Clin. Immunol. 2011, 127, 355–360. [Google Scholar] [CrossRef]
- Kim, H.J.; Jeong, J.S.; Kim, S.R.; Park, S.Y.; Chae, H.J.; Lee, Y.C. Inhibition of endoplasmic reticulum stress alleviates lipopolysaccharide-induced lung inflammation through modulation of NF-kappaB/HIF-1alpha signaling pathway. Sci. Rep. 2013, 3, 1142. [Google Scholar] [CrossRef]
- Bonnet, M.C.; Weil, R.; Dam, E.; Hovanessian, A.G.; Meurs, E.F. PKR stimulates NF-kappaB irrespective of its kinase function by interacting with the IkappaB kinase complex. Mol. Cell. Biol. 2000, 20, 4532–4542. [Google Scholar] [CrossRef]
- Cabanski, M.; Steinmuller, M.; Marsh, L.M.; Surdziel, E.; Seeger, W.; Lohmeyer, J. PKR regulates TLR2/TLR4-dependent signaling in murine alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 2008, 38, 26–31. [Google Scholar] [CrossRef]
- Ciencewicki, J.; Trivedi, S.; Kleeberger, S.R. Oxidants and the pathogenesis of lung diseases. J. Allergy Clin. Immunol. 2008, 122, 456–468; quiz 469–470. [Google Scholar] [CrossRef]
- Barnes, P.J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2013, 131, 636–645. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef]
- Kim, S.R.; Kim, D.I.; Kim, S.H.; Lee, H.; Lee, K.S.; Cho, S.H.; Lee, Y.C. NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis. 2014, 5, e1498. [Google Scholar] [CrossRef]
- Xu, W.; Ghosh, S.; Comhair, S.A.; Asosingh, K.; Janocha, A.J.; Mavrakis, D.A.; Bennett, C.D.; Gruca, L.L.; Graham, B.B.; Queisser, K.A.; et al. Increased mitochondrial arginine metabolism supports bioenergetics in asthma. J. Clin. Investig. 2016, 126, 2465–2481. [Google Scholar] [CrossRef]
- Cloonan, S.M.; Choi, A.M. Mitochondria in lung disease. J. Clin. Investig. 2016, 126, 809–820. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Besnard, A.G.; Guillou, N.; Tschopp, J.; Erard, F.; Couillin, I.; Iwakura, Y.; Quesniaux, V.; Ryffel, B.; Togbe, D. NLRP3 inflammasome is required in murine asthma in the absence of aluminum adjuvant. Allergy 2011, 66, 1047–1057. [Google Scholar] [CrossRef]
- Kool, M.; Willart, M.A.; van Nimwegen, M.; Bergen, I.; Pouliot, P.; Virchow, J.C.; Rogers, N.; Osorio, F.; Reis e Sousa, C.; Hammad, H.; et al. An unexpected role for uric acid as an inducer of T helper 2 cell immunity to inhaled antigens and inflammatory mediator of allergic asthma. Immunity 2011, 34, 527–540. [Google Scholar] [CrossRef]
- Kim, R.Y.; Pinkerton, J.W.; Essilfie, A.T.; Robertson, A.A.B.; Baines, K.J.; Brown, A.C.; Mayall, J.R.; Ali, M.K.; Starkey, M.R.; Hansbro, N.G.; et al. Role for NLRP3 Inflammasome-mediated, IL-1beta-Dependent Responses in Severe, Steroid-Resistant Asthma. Am. J. Respir. Crit. Care Med. 2017, 196, 283–297. [Google Scholar] [CrossRef]
- Simpson, J.L.; Phipps, S.; Baines, K.J.; Oreo, K.M.; Gunawardhana, L.; Gibson, P.G. Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur. Respir. J. 2014, 43, 1067–1076. [Google Scholar] [CrossRef]
- Jeong, J.S.; Lee, K.B.; Kim, S.R.; Kim, D.I.; Park, H.J.; Lee, H.K.; Kim, H.J.; Cho, S.H.; Kolliputi, N.; Kim, S.H.; et al. Airway epithelial phosphoinositide 3-kinase-delta contributes to the modulation of fungi-induced innate immune response. Thorax 2018, 73, 758–768. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Marwick, J.A.; Caramori, G.; Stevenson, C.S.; Casolari, P.; Jazrawi, E.; Barnes, P.J.; Ito, K.; Adcock, I.M.; Kirkham, P.A.; Papi, A. Inhibition of PI3Kdelta restores glucocorticoid function in smoking-induced airway inflammation in mice. Am. J. Respir. Crit. Care Med. 2009, 179, 542–548. [Google Scholar] [CrossRef]
- Bel, E.H.; Sousa, A.; Fleming, L.; Bush, A.; Chung, K.F.; Versnel, J.; Wagener, A.H.; Wagers, S.S.; Sterk, P.J.; Compton, C.H. Diagnosis and definition of severe refractory asthma: An international consensus statement from the Innovative Medicine Initiative (IMI). Thorax 2011, 66, 910–917. [Google Scholar] [CrossRef]
- Shaw, D.E.; Sousa, A.R.; Fowler, S.J.; Fleming, L.J.; Roberts, G.; Corfield, J.; Pandis, I.; Bansal, A.T.; Bel, E.H.; Auffray, C.; et al. Clinical and inflammatory characteristics of the European U-BIOPRED adult severe asthma cohort. Eur. Respir. J. 2015, 46, 1308–1321. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, J.S.; Kim, S.R.; Cho, S.H.; Lee, Y.C. A Novel Insight on Endotyping Heterogeneous Severe Asthma Based on Endoplasmic Reticulum Stress: Beyond the “Type 2/Non-Type 2 Dichotomy”. Int. J. Mol. Sci. 2019, 20, 713. https://doi.org/10.3390/ijms20030713
Jeong JS, Kim SR, Cho SH, Lee YC. A Novel Insight on Endotyping Heterogeneous Severe Asthma Based on Endoplasmic Reticulum Stress: Beyond the “Type 2/Non-Type 2 Dichotomy”. International Journal of Molecular Sciences. 2019; 20(3):713. https://doi.org/10.3390/ijms20030713
Chicago/Turabian StyleJeong, Jae Seok, So Ri Kim, Seong Ho Cho, and Yong Chul Lee. 2019. "A Novel Insight on Endotyping Heterogeneous Severe Asthma Based on Endoplasmic Reticulum Stress: Beyond the “Type 2/Non-Type 2 Dichotomy”" International Journal of Molecular Sciences 20, no. 3: 713. https://doi.org/10.3390/ijms20030713
APA StyleJeong, J. S., Kim, S. R., Cho, S. H., & Lee, Y. C. (2019). A Novel Insight on Endotyping Heterogeneous Severe Asthma Based on Endoplasmic Reticulum Stress: Beyond the “Type 2/Non-Type 2 Dichotomy”. International Journal of Molecular Sciences, 20(3), 713. https://doi.org/10.3390/ijms20030713