Parkin Interacts with Apoptosis-Inducing Factor and Interferes with Its Translocation to the Nucleus in Neuronal Cells


 , , and
, , and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
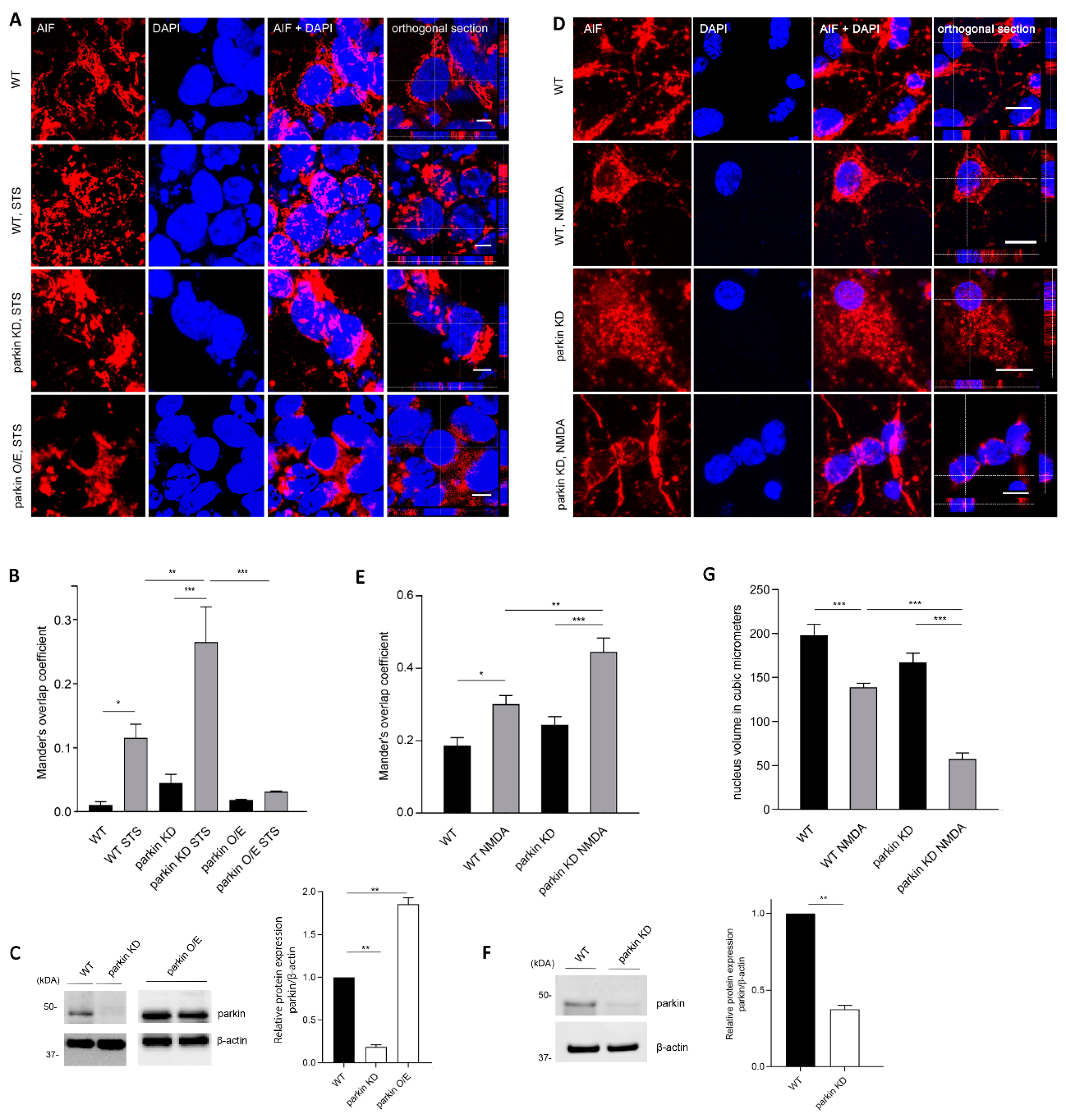
2.1. Parkin Interacts with AIF
2.2. Parkin Reduces AIF Translocation to the Nucleus
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Immunostaining and Proximity Ligation Assay
4.3. Co-Immunoprecipitation and Western Blot
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AIF | apoptosis-inducing factor |
| AIMP2 | aminoacyl-tRNA synthetase complex-interacting multifunctional protein-2 |
| ANOVA | one-way analysis of variance |
| CCCP | carbonyl cyanide m-chlorophenylhydrazone |
| Co-IP | co-immunoprecipitation |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s modified Eagle’s Medium |
| DMSO | Dimethyl sulfoxide |
| DIV | Days in vitro |
| GFP | green fluorescent protein |
| HA | Hemagglutinin |
| IP | Immunoprecipitation |
| kDA | kilodaltons |
| MIF | macrophage migration inhibitory factor |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NMDA | N-methyl-d-aspartate |
| PAR | poly ADP-ribose |
| PARP1 | Poly(ADP-ribose) polymerase-1 |
| PD | Parkinson’s disease |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator-1 alpha |
| PINK1 | PTEN induced kinase 1 |
| PLA | Proximity Ligation Assay |
| PRKN | Parkin |
| STS | Staurosporine |
| WB | Western blot |
| WT | Wild type |
References
- Klein, C.; Lohmann-Hedrich, K. Impact of recent genetic findings in Parkinson’s disease. Curr. Opin. Neurol. 2007, 20, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Dawson, V.L. Parkin plays a role in sporadic Parkinson’s disease. Neurodegener. Dis. 2014, 13, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ren, Y.; Zhao, J.; Feng, J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum. Mol. Genet. 2004, 13, 1745–1754. [Google Scholar] [CrossRef] [Green Version]
- Vercammen, L.; Van der Perren, A.; Vaudano, E.; Gijsbers, R.; Debyser, Z.; Van den Haute, C.; Baekelandt, V. Parkin protects against neurotoxicity in the 6-hydroxydopamine rat model for Parkinson’s disease. Mol. Ther. 2006, 14, 716–723. [Google Scholar] [CrossRef]
- Whitworth, A.J.; Theodore, D.A.; Greene, J.C.; Benes, H.; Wes, P.D.; Pallanck, L.J. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 8024–8029. [Google Scholar] [CrossRef] [PubMed]
- Bouman, L.; Schlierf, A.; Lutz, A.K.; Shan, J.; Deinlein, A.; Kast, J.; Galehdar, Z.; Palmisano, V.; Patenge, N.; Berg, D.; et al. Parkin is transcriptionally regulated by ATF4: Evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011, 18, 769–782. [Google Scholar] [CrossRef]
- Imai, Y.; Soda, M.; Takahashi, R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 2000, 275, 35661–35664. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2010, 25 (Suppl. 1), S32–S39. [Google Scholar] [CrossRef]
- Rothfuss, O.; Fischer, H.; Hasegawa, T.; Maisel, M.; Leitner, P.; Miesel, F.; Sharma, M.; Bornemann, A.; Berg, D.; Gasser, T.; et al. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum. Mol. Genet. 2009, 18, 3832–3850. [Google Scholar] [CrossRef] [Green Version]
- Darios, F.; Corti, O.; Lucking, C.B.; Hampe, C.; Muriel, M.P.; Abbas, N.; Gu, W.J.; Hirsch, E.C.; Rooney, T.; Ruberg, M.; et al. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum. Mol. Genet. 2003, 12, 517–526. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Wauer, T.; Simicek, M.; Schubert, A.; Komander, D. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 2015, 524, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Gladkova, C.; Maslen, S.L.; Skehel, J.M.; Komander, D. Mechanism of parkin activation by PINK1. Nature 2018, 559, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Muller-Rischart, A.K.; Pilsl, A.; Beaudette, P.; Patra, M.; Hadian, K.; Funke, M.; Peis, R.; Deinlein, A.; Schweimer, C.; Kuhn, P.H.; et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol. Cell 2013, 49, 908–921. [Google Scholar] [CrossRef]
- Charan, R.A.; Johnson, B.N.; Zaganelli, S.; Nardozzi, J.D.; LaVoie, M.J. Inhibition of apoptotic Bax translocation to the mitochondria is a central function of parkin. Cell Death Dis. 2014, 5, e1313. [Google Scholar] [CrossRef]
- Fallon, L.; Moreau, F.; Croft, B.G.; Labib, N.; Gu, W.J.; Fon, E.A. Parkin and CASK/LIN-2 associate via a PDZ-mediated interaction and are co-localized in lipid rafts and postsynaptic densities in brain. J. Biol. Chem. 2002, 277, 486–491. [Google Scholar] [CrossRef]
- Imai, Y.; Soda, M.; Hatakeyama, S.; Akagi, T.; Hashikawa, T.; Nakayama, K.I.; Takahashi, R. CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol. Cell 2002, 10, 55–67. [Google Scholar] [CrossRef]
- Smith, W.W.; Pei, Z.; Jiang, H.; Moore, D.J.; Liang, Y.; West, A.B.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 18676–18681. [Google Scholar] [CrossRef] [Green Version]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Daugas, E.; Susin, S.A.; Zamzami, N.; Ferri, K.F.; Irinopoulou, T.; Larochette, N.; Prevost, M.C.; Leber, B.; Andrews, D.; Penninger, J.; et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000, 14, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Joza, N.; Steenaart, N.A.; McClellan, K.A.; Neuspiel, M.; McNamara, S.; MacLaurin, J.G.; Rippstein, P.; Park, D.S.; Shore, G.C.; et al. Dissociating the dual roles of apoptosis-inducing factor in maintaining mitochondrial structure and apoptosis. EMBO J. 2006, 25, 4061–4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otera, H.; Ohsakaya, S.; Nagaura, Z.; Ishihara, N.; Mihara, K. Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J. 2005, 24, 1375–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moubarak, R.S.; Yuste, V.J.; Artus, C.; Bouharrour, A.; Greer, P.A.; Menissier-de Murcia, J.; Susin, S.A. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol. Cell Biol. 2007, 27, 4844–4862. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, J.; Graham, S.H.; Du, L.; Kochanek, P.M.; Draviam, R.; Guo, F.; Nathaniel, P.D.; Szabo, C.; Watkins, S.C.; et al. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J. Neurochem. 2002, 82, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Clark, R.S.; Pei, W.; Yin, W.; Zhang, F.; Sun, F.Y.; Graham, S.H.; Chen, J. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J. Cereb Blood Flow Metab. 2003, 23, 1137–1150. [Google Scholar] [CrossRef]
- Burguillos, M.A.; Hajji, N.; Englund, E.; Persson, A.; Cenci, A.M.; Machado, A.; Cano, J.; Joseph, B.; Venero, J.L. Apoptosis-inducing factor mediates dopaminergic cell death in response to LPS-induced inflammatory stimulus: evidence in Parkinson’s disease patients. Neurobiol. Dis. 2011, 41, 177–188. [Google Scholar] [CrossRef]
- Chu, C.T.; Zhu, J.H.; Cao, G.; Signore, A.; Wang, S.; Chen, J. Apoptosis inducing factor mediates caspase-independent 1-methyl-4-phenylpyridinium toxicity in dopaminergic cells. J. Neurochem. 2005, 94, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Yalcinkaya, N.; Haytural, H.; Bilgic, B.; Ozdemir, O.; Hanagasi, H.; Kucukali, C.I.; Ozbek, Z.; Akcan, U.; Idrisoglu, H.A.; Gurvit, H.; et al. Expression changes of genes associated with apoptosis and survival processes in Parkinson’s disease. Neurosci. Lett. 2016, 615, 72–77. [Google Scholar] [CrossRef]
- Peneder, T.M.; Bauer, J.; Pifl, C. Apoptosis-inducing factor in nigral dopamine neurons: Higher levels in primates than in mice. Mov. Disord. 2016, 31, 1729–1733. [Google Scholar] [CrossRef] [PubMed]
- Zanon, A.; Rakovic, A.; Blankenburg, H.; Doncheva, N.T.; Schwienbacher, C.; Serafin, A.; Alexa, A.; Weichenberger, C.X.; Albrecht, M.; Klein, C.; et al. Profiling of Parkin-Binding Partners Using Tandem Affinity Purification. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Belmokhtar, C.A.; Hillion, J.; Segal-Bendirdjian, E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 2001, 20, 3354–3362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.W.; Wang, Y.; Frydenlund, D.S.; Ottersen, O.P.; Dawson, V.L.; Dawson, T.M. Outer mitochondrial membrane localization of apoptosis-inducing factor: mechanistic implications for release. ASN Neuro. 2009, 1. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann. N. Y. Acad. Sci. 2008, 1147, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Kim, N.S.; Haince, J.F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 2011, 4, ra20. [Google Scholar] [CrossRef]
- Wang, Y.; An, R.; Umanah, G.K.; Park, H.; Nambiar, K.; Eacker, S.M.; Kim, B.; Bao, L.; Harraz, M.M.; Chang, C.; et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 2016, 354. [Google Scholar] [CrossRef]
- Carroll, R.G.; Hollville, E.; Martin, S.J. Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep. 2014, 9, 1538–1553. [Google Scholar] [CrossRef]
- Zhang, C.; Lee, S.; Peng, Y.; Bunker, E.; Giaime, E.; Shen, J.; Zhou, Z.; Liu, X. PINK1 triggers autocatalytic activation of Parkin to specify cell fate decisions. Curr. Biol. 2014, 24, 1854–1865. [Google Scholar] [CrossRef]
- Le Guerroue, F.; Youle, R.J. Active state of Parkin. Nat. Struct. Mol. Biol. 2018, 25, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Sauve, V.; Sung, G.; Soya, N.; Kozlov, G.; Blaimschein, N.; Miotto, L.S.; Trempe, J.F.; Lukacs, G.L.; Gehring, K. Mechanism of parkin activation by phosphorylation. Nat. Struct. Mol. Biol. 2018, 25, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Karuppagounder, S.S.; Shin, J.H.; Lee, Y.I.; Ko, H.S.; Swing, D.; Jiang, H.; Kang, S.U.; Lee, B.D.; Kang, H.C.; et al. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci. 2013, 16, 1392–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, S.; Costa, A.C.; Celardo, I.; Loh, S.H.; Martins, L.M. Parp mutations protect against mitochondrial dysfunction and neurodegeneration in a PARKIN model of Parkinson’s disease. Cell Death Dis. 2016, 7, e2166. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.Y.; Kishinevsky, S.; Mazzulli, J.R.; Graziotto, J.; Mrejeru, A.; Mosharov, E.V.; Puspita, L.; Valiulahi, P.; Sulzer, D.; Milner, T.A.; et al. Parkin and PINK1 Patient iPSC-Derived Midbrain Dopamine Neurons Exhibit Mitochondrial Dysfunction and alpha-Synuclein Accumulation. Stem Cell Rep. 2016, 7, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Bondada, V.; Geddes, J.W. Mitochondrial micro-calpain is not involved in the processing of apoptosis-inducing factor. Exp. Neurol. 2009, 218, 221–227. [Google Scholar] [CrossRef]
- Zanon, A.; Kalvakuri, S.; Rakovic, A.; Foco, L.; Guida, M.; Schwienbacher, C.; Serafin, A.; Rudolph, F.; Trilck, M.; Grunewald, A.; et al. SLP-2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in Parkin-deficient human iPSC-derived neurons and Drosophila. Hum. Mol. Genet. 2017, 26, 2412–2425. [Google Scholar] [CrossRef]
- Pischedda, F.; Montani, C.; Obergasteiger, J.; Frapporti, G.; Corti, C.; Rosato Siri, M.; Volta, M.; Piccoli, G. Cryopreservation of Primary Mouse Neurons: The Benefit of Neurostore Cryoprotective Medium. Front. Cell Neurosci. 2018, 12, 81. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guida, M.; Zanon, A.; Montibeller, L.; Lavdas, A.A.; Ladurner, J.; Pischedda, F.; Rakovic, A.; Domingues, F.S.; Piccoli, G.; Klein, C.; et al. Parkin Interacts with Apoptosis-Inducing Factor and Interferes with Its Translocation to the Nucleus in Neuronal Cells. Int. J. Mol. Sci. 2019, 20, 748. https://doi.org/10.3390/ijms20030748
Guida M, Zanon A, Montibeller L, Lavdas AA, Ladurner J, Pischedda F, Rakovic A, Domingues FS, Piccoli G, Klein C, et al. Parkin Interacts with Apoptosis-Inducing Factor and Interferes with Its Translocation to the Nucleus in Neuronal Cells. International Journal of Molecular Sciences. 2019; 20(3):748. https://doi.org/10.3390/ijms20030748
Chicago/Turabian StyleGuida, Marianna, Alessandra Zanon, Luigi Montibeller, Alexandros A. Lavdas, Judith Ladurner, Francesca Pischedda, Aleksandar Rakovic, Francisco S. Domingues, Giovanni Piccoli, Christine Klein, and et al. 2019. "Parkin Interacts with Apoptosis-Inducing Factor and Interferes with Its Translocation to the Nucleus in Neuronal Cells" International Journal of Molecular Sciences 20, no. 3: 748. https://doi.org/10.3390/ijms20030748
APA StyleGuida, M., Zanon, A., Montibeller, L., Lavdas, A. A., Ladurner, J., Pischedda, F., Rakovic, A., Domingues, F. S., Piccoli, G., Klein, C., Pramstaller, P. P., Hicks, A. A., & Pichler, I. (2019). Parkin Interacts with Apoptosis-Inducing Factor and Interferes with Its Translocation to the Nucleus in Neuronal Cells. International Journal of Molecular Sciences, 20(3), 748. https://doi.org/10.3390/ijms20030748