Is Retinal Metabolic Dysfunction at the Center of the Pathogenesis of Age-related Macular Degeneration?


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
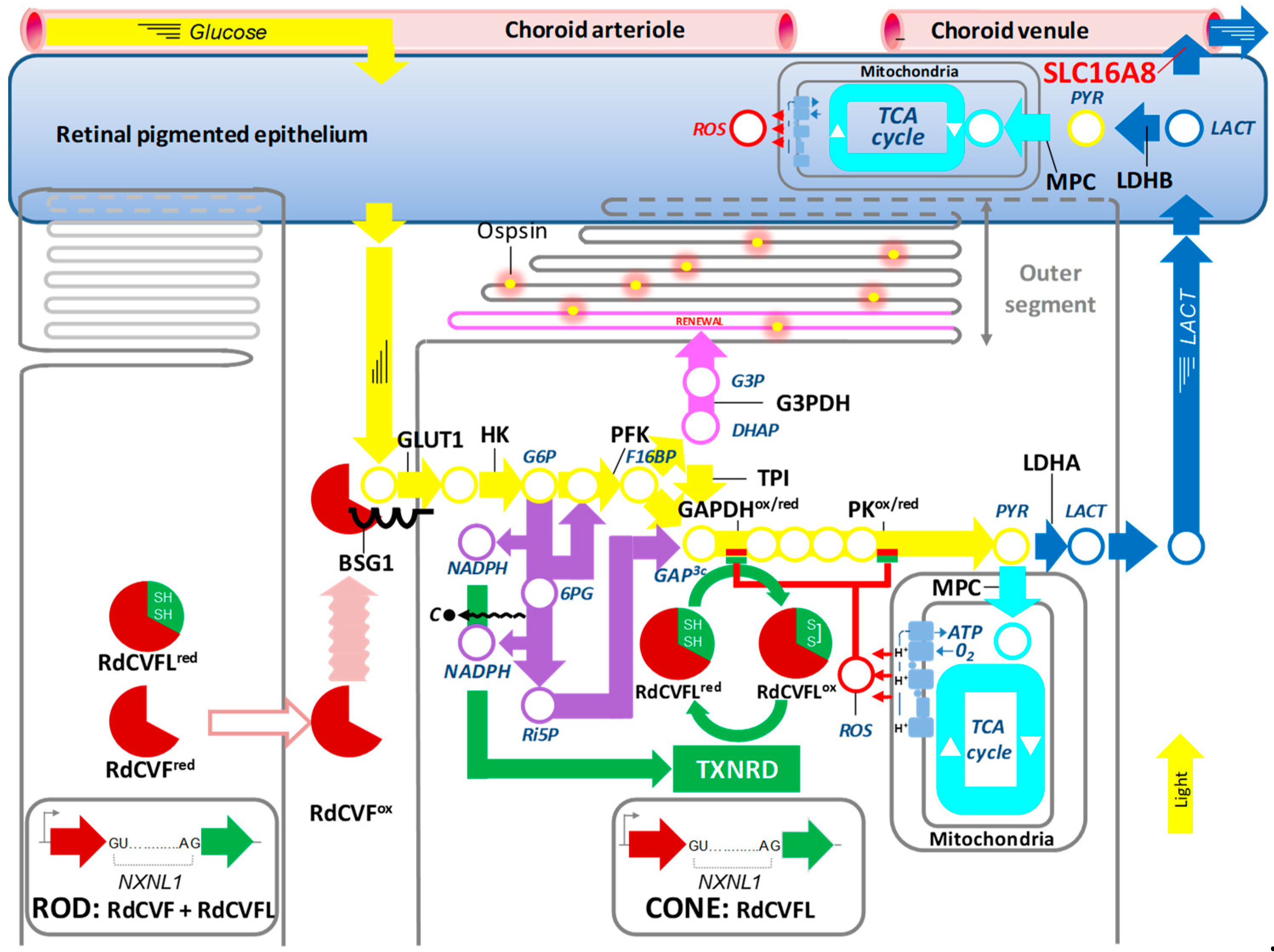
2. Photoreceptor Metabolism and Rod-derived Cone Viability Factor
3. Age-related Macular Degeneration
4. Chronic Low-Grade Inflammation in AMD
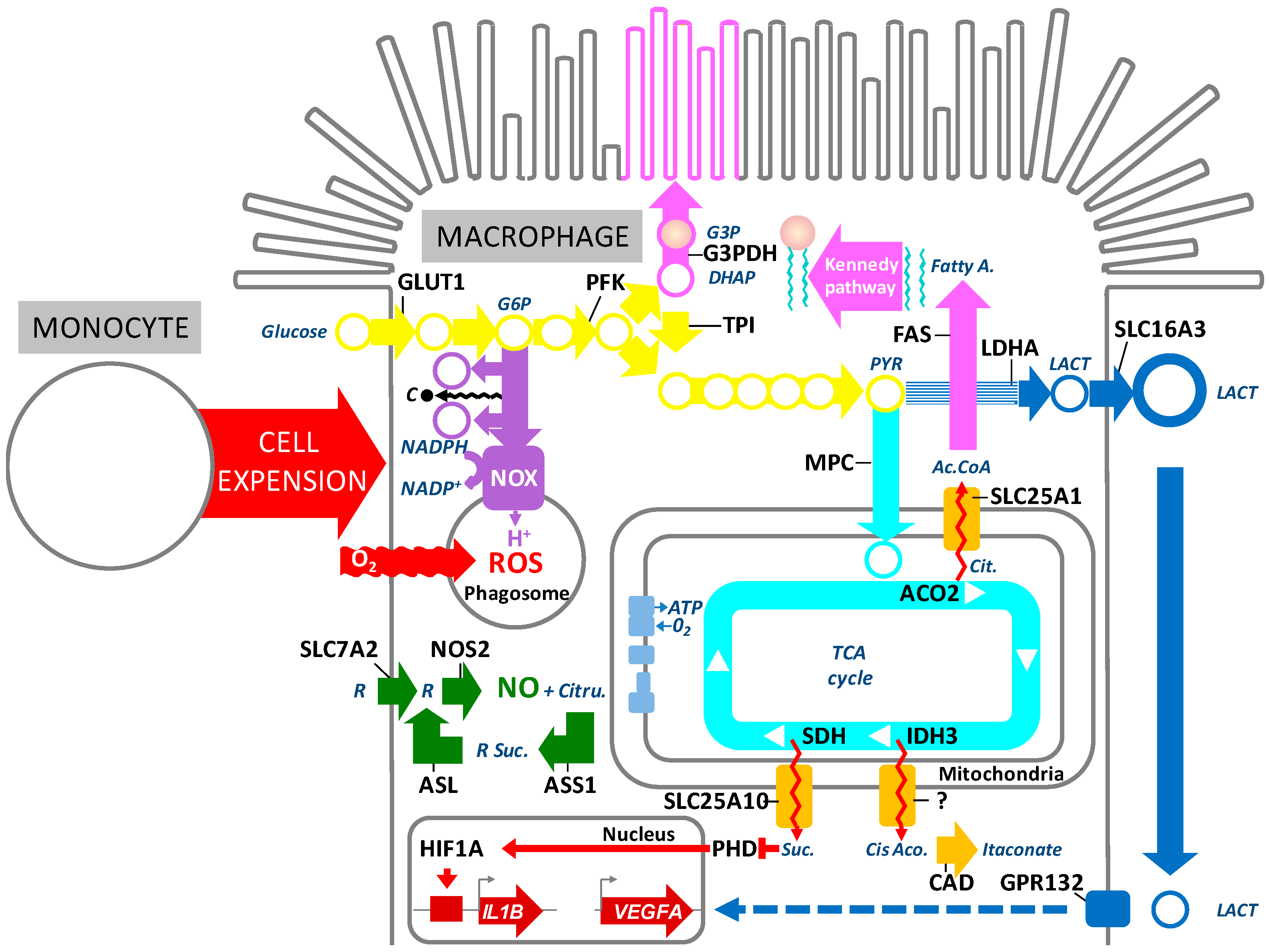
5. Inflammation-Induced Metabolic Changes in AMD
6. Genetic Susceptibility for Subretinal Metabolic Changes
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 6PG | 6-phosphogluconate |
| Acetyl-CoA | Acetyl-coenzyme A |
| ACO2 | Aconitase mitochondrial |
| ALDO | Aldolase |
| AMD | Age-related macular degeneration |
| APO E | Apolipoprotein E |
| ASL | Argininosuccinate lyase |
| ASS1 | Argininosuccinate synthase 1 |
| ATP | Adenosine triphosphate |
| BSG | Basigin |
| BSG1 | Basigin splice variant 1 |
| BSG2 | Basigin splice variant 2 |
| CAD | Cis-aconitate decarboxylase |
| CCL2 | C–C motif chemokine 2 |
| CCR2 | CCL2 receptor |
| CD | Cell determinant |
| CFH | Complement factor H |
| CNV | Choroid neovascularization |
| CX3CR1 | CX3C chemokine receptor 1 |
| DHAP | Dihydroxyacetone phosphate |
| F16BP | Fructose-1,6-biphosphate |
| G3P | Glycerol-3-phosphate |
| G3PDH | Glycerol-3-phosphate dehydrogenase |
| G6P | Glucose-6-phosphate |
| GA | Geographic atrophy |
| GAP | Glyceraldyde-3-phosphate |
| GAPDH | Glyceraldehyde-3 phosphate-dehydrogenase |
| GLUT1 | Glucose transporter 1 |
| GPR91 | G-protein coupled receptor 91 (Succinate receptor 1) |
| GPR132 | G-protein coupled receptor 132 |
| HIF1A | Hypoxia-inducible factor 1 |
| IDH3 | Isocitrate dehydrogenase 3 |
| Ig | Immunoglobulin |
| IL1B | Interleukin-1β |
| IL6 | Interleukin-6 |
| IRG1 | Immune-responsive gene 1 |
| LACT | Lactate |
| LDHA | Lactate dehydrogenase A |
| LDHB | Lactate dehydrogenase B |
| MPC | Mitochondrial pyruvate carrier |
| NADH | Nicotinamide adenine dinucleotide (reduced) |
| NADP+ | Nicotinamide adenine dinucleotide phosphate (oxidized) |
| NADPH | Nicotinamide adenine dinucleotide phosphate (reduced) |
| NOS2 | Inducible nitric oxide synthase |
| NOX | NADPH oxidase |
| NXNL1 | Nucleoredoxin-like 1 |
| OXPHO | Oxidative phosphorylation |
| PHD | Prolyl-hydroxylase |
| PK | Pyruvate kinase |
| PPP | Pentose phosphate pathway |
| PUFA | Poly-unsaturated fatty acids |
| PYR | Pyruvate |
| RdCVF | Rod-derived cone viability factor |
| RdCVFL | Rod-derived cone viability factor long (thioredoxin) |
| RdCVFox | Rod-derived cone viability factor with oxidized cysteines |
| Ri5P | Ribulose-5-phosphate |
| ROS | Reactive oxygen species |
| RPE | Retinal pigmented epithelium |
| SCR7 | Short tandem repeat 7 |
| SDH | Succinate dehydrogenase |
| SLC16A1 | Monocarboxylate transporter 1 (MCT1) |
| SLC16A3 | Monocarboxylate transporter 4 (MCT4) |
| SLC16A8 | Monocarboxylate transporter 3 (MCT3) |
| SLC25A10 | Mitochondrial dicarboxylate carrier |
| SLC2A1 | Glucose transporter 1 (GLUT1) |
| SLC7A2 | Arginine transporter |
| TCA | Tricarboxylic acid |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor-α |
| TNFSF6 | FAS ligand |
| TPI | Triose phosphate isomerase |
| TSP1 | Thrombospondin 1 |
| TXNRD | Thioredoxin reductase |
| VEGF | Vascular endothelial growth factor |
References
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; W.H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Leveillard, T. Cancer metabolism of cone photoreceptors. Oncotarget 2015, 6, 32285–32286. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Nonnenmacher, Y.; Hiller, K. Biochemistry of proinflammatory macrophage activation. Cell. Mol. Life Sci. 2018, 75, 2093–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audo, I.; Bujakowska, K.; Orhan, E.; El Shamieh, S.; Sennlaub, F.; Guillonneau, X.; Antonio, A.; Michiels, C.; Lancelot, M.E.; Letexier, M.; et al. The familial dementia gene revisited: A missense mutation revealed by whole-exome sequencing identifies ITM2B as a candidate gene underlying a novel autosomal dominant retinal dystrophy in a large family. Human Mol. Gen. 2014, 23, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Calippe, B.; Augustin, S.; Beguier, F.; Charles-Messance, H.; Poupel, L.; Conart, J.B.; Hu, S.J.; Lavalette, S.; Fauvet, A.; Rayes, J.; et al. Complement Factor H Inhibits CD47-Mediated Resolution of Inflammation. Immunity 2017, 46, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Rubartelli, A.; Bajetto, A.; Allavena, G.; Wollman, E.; Sitia, R. Secretion of Thioredoxin by Normal and Neoplastic-Cells through a Leaderless Secretory Pathway. J. Biol. Chem. 1992, 267, 24161–24164. [Google Scholar] [PubMed]
- Ait-Ali, N.; Fridlich, R.; Millet-Puel, G.; Clerin, E.; Delalande, F.; Jaillard, C.; Blond, F.; Perrocheau, L.; Reichman, S.; Byrne, L.C.; et al. Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Cell 2015, 161, 817–832. [Google Scholar] [CrossRef]
- Ochrietor, J.D.; Moroz, T.P.; van Ekeris, L.; Clamp, M.F.; Jefferson, S.C.; deCarvalho, A.C.; Fadool, J.M.; Wistow, G.; Muramatsu, T.; Linser, P.J. Retina-specific expression of 5A11/Basigin-2, a member of the immunoglobulin gene superfamily. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4086–4096. [Google Scholar] [CrossRef]
- Camacho, E.T.; Leveillard, T.; Sahel, J.A.; Wirkus, S. Mathematical Model of the Role of RdCVF in the Coexistence of Rods and Cones in a Healthy Eye. Bull. Math. Biol. 2016, 78, 1394–1409. [Google Scholar] [CrossRef]
- Hebert, D.N.; Carruthers, A. Glucose transporter oligomeric structure determines transporter function. Reversible redox-dependent interconversions of tetrameric and dimeric GLUT1. J. Biol. Chem. 1992, 267, 23829–23838. [Google Scholar] [PubMed]
- Soares Moretti, A.I.; Martins Laurindo, F.R. Protein disulfide isomerases: Redox connections in and out of the endoplasmic reticulum. Arch. Biochem. Biophys. 2017, 617, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Punzo, C.; Kornacker, K.; Cepko, C.L. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat. Neurosci. 2009, 12, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Rajala, A.; Wang, Y.; Rajala, R.V. Activation of oncogenic tyrosine kinase signaling promotes insulin receptor-mediated cone photoreceptor survival. Oncotarget 2016, 7, 46924–46942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinchore, Y.; Begaj, T.; Wu, D.; Drokhlyansky, E.; Cepko, C.L. Glycolytic reliance promotes anabolism in photoreceptors. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adijanto, J.; Philp, N.J. The SLC16A family of monocarboxylate transporters (MCTs)—Physiology and function in cellular metabolism, pH homeostasis, and fluid transport. Curr. Topics Membr. 2012, 70, 275–311. [Google Scholar]
- Kanow, M.A.; Giarmarco, M.M.; Jankowski, C.S.; Tsantilas, K.; Engel, A.L.; Du, J.; Linton, J.D.; Farnsworth, C.C.; Sloat, S.R.; Rountree, A.; et al. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, G.L.; Benedicto, I.; Philp, N.J.; Rodriguez-Boulan, E. Plasma membrane protein polarity and trafficking in RPE cells: Past, present and future. Exp. Eye Res. 2014, 126, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Daniele, L.L.; Sauer, B.; Gallagher, S.M.; Pugh, E.N., Jr.; Philp, N.J. Altered visual function in monocarboxylate transporter 3 (Slc16a8) knockout mice. Am. J. Physiol. Cell Physiol. 2008, 295, C451–C457. [Google Scholar] [CrossRef] [Green Version]
- Mei, X.; Chaffiol, A.; Kole, C.; Yang, Y.; Millet-Puel, G.; Clerin, E.; Ait-Ali, N.; Bennett, J.; Dalkara, D.; Sahel, J.A.; et al. The Thioredoxin Encoded by the Rod-Derived Cone Viability Factor Gene Protects Cone Photoreceptors Against Oxidative Stress. Antioxid Redox Signal 2016, 24, 909–923. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Fridlich, R.; Delalande, F.; Jaillard, C.; Lu, J.; Poidevin, L.; Cronin, T.; Perrocheau, L.; Millet-Puel, G.; Niepon, M.L.; Poch, O.; et al. The thioredoxin-like protein rod-derived cone viability factor (RdCVFL) interacts with TAU and inhibits its phosphorylation in the retina. Mol. Cell. Proteom. 2009, 8, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.; Maslim, J.; Valter-Kocsi, K.; Mervin, K.; Bowers, F.; Chu, Y.; Barnett, N.; Provis, J.; Lewis, G.; Fisher, S.K.; et al. Mechanisms of photoreceptor death and survival in mammalian retina. Prog. Retin. Eye Res. 1999, 18, 689–735. [Google Scholar] [CrossRef]
- Roberts, P.A.; Gaffney, E.A.; Luthert, P.J.; Foss, A.J.E.; Byrne, H.M. Mathematical models of retinitis pigmentosa: The oxygen toxicity hypothesis. J. Theor. Biol. 2017, 425, 53–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komeima, K.; Rogers, B.S.; Lu, L.; Campochiaro, P.A. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2006, 103, 11300–11305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busskamp, V.; Duebel, J.; Balya, D.; Fradot, M.; Viney, T.J.; Siegert, S.; Groner, A.C.; Cabuy, E.; Forster, V.; Seeliger, M.; et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science 2010, 329, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lee, S.J.; Scott, P.A.; Lu, X.; Emery, D.; Liu, Y.; Ezashi, T.; Roberts, M.R.; Ross, J.W.; Kaplan, H.J.; et al. Two-Step Reactivation of Dormant Cones in Retinitis Pigmentosa. Cell Rep. 2016, 15, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.C.; Parker, A.B.; Botelho, J.V.; Duncan, J.L. Spontaneous Regeneration of Human Photoreceptor Outer Segments. Sci. Rep. 2015, 5, 12364. [Google Scholar] [CrossRef] [Green Version]
- Guillonneau, X.; Eandi, C.M.; Paques, M.; Sahel, J.A.; Sapieha, P.; Sennlaub, F. On phagocytes and macular degeneration. Prog. Retin. Eye Res. 2017, 61, 98–128. [Google Scholar] [CrossRef] [Green Version]
- De Jong, P.T.V.M. Elusive drusen and changing terminology of AMD. Eye 2018, 32, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardeljan, D.; Chan, C.C. Aging is not a disease: Distinguishing age-related macular degeneration from aging. Prog. Ret. Eye Res. 2013, 37, 68–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.; Peto, T.; Bird, A.; Vannewkirk, M.R. The epidemiology of age-related macular degeneration. Am. J. Ophthalmol. 2004, 137, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Foran, S.; Smith, W.; Mitchell, P. Risk of age-related macular degeneration in eyes with macular drusen or hyperpigmentation: The Blue Mountains Eye Study cohort. Arch. Ophthalmol. 2003, 121, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Mimoun, G.; Soubrane, G.; Coscas, G. Macular drusen. Journal Francais D’ophtalmologie 1990, 13, 511–530. [Google Scholar] [PubMed]
- Klein, R.; Meuer, S.M.; Knudtson, M.D.; Iyengar, S.K.; Klein, B.E. The epidemiology of retinal reticular drusen. Am. J. Ophthalmol. 2008, 145, 317–326. [Google Scholar] [CrossRef]
- Rudolf, M.; Malek, G.; Messinger, J.D.; Clark, M.E.; Wang, L.; Curcio, C.A. Sub-retinal drusenoid deposits in human retina: Organization and composition. Exp. Eye Res. 2008, 87, 402–408. [Google Scholar] [CrossRef] [Green Version]
- Zweifel, S.A.; Spaide, R.F.; Curcio, C.A.; Malek, G.; Imamura, Y. Reticular Pseudodrusen Are Subretinal Drusenoid Deposits. Ophthalmology 2010, 117, 303–312. [Google Scholar] [CrossRef]
- Alten, F.; Eter, N. Current knowledge on reticular pseudodrusen in age-related macular degeneration. Br. J. Ophthalmol. 2015, 99, 717–722. [Google Scholar] [CrossRef]
- Sarks, S.H. Ageing and degeneration in the macular region: A clinico-pathological study. Br. J. Ophthalmol. 1976, 60, 324–341. [Google Scholar] [CrossRef]
- Bird, A.C.; Phillips, R.L.; Hageman, G.S. Geographic atrophy: A histopathological assessment. JAMA Ophthalmol. 2014, 132, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Eandi, C.M.; Charles Messance, H.; Augustin, S.; Dominguez, E.; Lavalette, S.; Forster, V.; Hu, S.J.; Siquieros, L.; Craft, C.M.; Sahel, J.A.; et al. Subretinal mononuclear phagocytes induce cone segment loss via IL-1beta. eLife 2016, 5, e16490. [Google Scholar] [CrossRef] [PubMed]
- Curcio, C.A. Photoreceptor topography in ageing and age-related maculopathy. Eye (Lond) 2001, 15 Pt 3, 376–383. [Google Scholar] [CrossRef]
- Meleth, A.D.; Mettu, P.; Agron, E.; Chew, E.Y.; Sadda, S.R.; Ferris, F.L.; Wong, W.T. Changes in retinal sensitivity in geographic atrophy progression as measured by microperimetry. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, Y.; Mitamura-Aizawa, S.; Katome, T.; Naito, T.; Hagiwara, A.; Kumagai, K.; Yamamoto, S. Photoreceptor impairment and restoration on optical coherence tomographic image. J. Ophthalmol. 2013, 2013, 518170. [Google Scholar] [CrossRef] [PubMed]
- Flamendorf, J.; Agron, E.; Wong, W.T.; Thompson, D.; Wiley, H.E.; Doss, E.L.; Al-Holou, S.; Ferris, F.L., 3rd; Chew, E.Y.; Cukras, C. Impairments in Dark Adaptation Are Associated with Age-Related Macular Degeneration Severity and Reticular Pseudodrusen. Ophthalmology 2015, 122, 2053–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owsley, C.; Jackson, G.R.; White, M.; Feist, R.; Edwards, D. Delays in rod-mediated dark adaptation in early age-related maculopathy. Ophthalmology 2001, 108, 1196–1202. [Google Scholar] [CrossRef]
- Owsley, C.; McGwin, G., Jr.; Clark, M.E.; Jackson, G.R.; Callahan, M.A.; Kline, L.B.; Witherspoon, C.D.; Curcio, C.A. Delayed Rod-Mediated Dark Adaptation Is a Functional Biomarker for Incident Early Age-Related Macular Degeneration. Ophthalmology 2016, 123, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Chirco, K.R.; Sohn, E.H.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Structural and molecular changes in the aging choroid: Implications for age-related macular degeneration. Eye (Lond) 2017, 31, 10–25. [Google Scholar] [CrossRef]
- Ghazi, N.G. Retinal angiomatous proliferation in age-related macular degeneration. Retina 2002, 22, 509–511, author reply 512. [Google Scholar] [CrossRef]
- Yannuzzi, L.A.; Negrao, S.; Iida, T.; Carvalho, C.; Rodriguez-Coleman, H.; Slakter, J.; Freund, K.B.; Sorenson, J.; Orlock, D.; Borodoker, N. Retinal angiomatous proliferation in age-related macular degeneration. Retina 2001, 21, 416–434. [Google Scholar] [CrossRef] [PubMed]
- McLeod, D.S.; Grebe, R.; Bhutto, I.; Merges, C.; Baba, T.; Lutty, G.A. Relationship between RPE and choriocapillaris in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4982–4991. [Google Scholar] [CrossRef] [PubMed]
- Rofagha, S.; Bhisitkul, R.B.; Boyer, D.S.; Sadda, S.R.; Zhang, K.; Group, S.-U.S. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: A multicenter cohort study (SEVEN-UP). Ophthalmology 2013, 120, 2292–2299. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadok, V.A.; Bratton, D.L.; Henson, P.M. Phagocyte receptors for apoptotic cells: Recognition, uptake, and consequences. J. Clin. Investig. 2001, 108, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.L.; Fadok, V.A.; Henson, P.M. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Investig. 2002, 109, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Gautier, E.L.; Ivanov, S.; Lesnik, P.; Randolph, G.J. Local apoptosis mediates clearance of macrophages from resolving inflammation in mice. Blood 2013, 122, 2714–2722. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef]
- Levy, O.; Calippe, B.; Lavalette, S.; Hu, S.J.; Raoul, W.; Dominguez, E.; Housset, M.; Paques, M.; Sahel, J.A.; Bemelmans, A.P.; et al. Apolipoprotein E promotes subretinal mononuclear phagocyte survival and chronic inflammation in age-related macular degeneration. EMBO Mol. Med. 2015, 7, 211–226. [Google Scholar] [CrossRef] [Green Version]
- Sennlaub, F.; Auvynet, C.; Calippe, B.; Lavalette, S.; Poupel, L.; Hu, S.J.; Dominguez, E.; Camelo, S.; Levy, O.; Guyon, E.; et al. CCR2(+) monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol. Med. 2013, 5, 1775–1793. [Google Scholar] [CrossRef]
- Combadiere, C.; Feumi, C.; Raoul, W.; Keller, N.; Rodero, M.; Pezard, A.; Lavalette, S.; Houssier, M.; Jonet, L.; Picard, E.; et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J. Clin. Investig. 2007, 117, 2920–2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.; Takagi, H.; Takagi, C.; Suzuma, K.; Otani, A.; Ishida, K.; Matsumura, M.; Ogura, Y.; Honda, Y. The potential angiogenic role of macrophages in the formation of choroidal neovascular membranes. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1891–1898. [Google Scholar] [PubMed]
- Jonas, J.B.; Tao, Y.; Neumaier, M.; Findeisen, P. Monocyte chemoattractant protein 1, intercellular adhesion molecule 1, and vascular cell adhesion molecule 1 in exudative age-related macular degeneration. Arch. Ophthalmol. 2010, 128, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Cousins, S.W.; Espinosa-Heidmann, D.G.; Csaky, K.G. Monocyte activation in patients with age-related macular degeneration: A biomarker of risk for choroidal neovascularization? Arch. Ophthalmol. 2004, 122, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Yanagi, Y.; Matsuura, K.; Takahashi, H.; Tamaki, Y.; Araie, M. Expression of hypoxia-inducible factor 1alpha and 2alpha in choroidal neovascular membranes associated with age-related macular degeneration. Br. J. Ophthalmol. 2007, 91, 1720–1721. [Google Scholar] [CrossRef]
- Tsutsumi, C.; Sonoda, K.H.; Egashira, K.; Qiao, H.; Hisatomi, T.; Nakao, S.; Ishibashi, M.; Charo, I.F.; Sakamoto, T.; Murata, T.; et al. The critical role of ocular-infiltrating macrophages in the development of choroidal neovascularization. J. Leukoc. Biol. 2003, 74, 25–32. [Google Scholar] [CrossRef]
- Sakurai, E.; Anand, A.; Ambati, B.K.; van Rooijen, N.; Ambati, J. Macrophage depletion inhibits experimental choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3578–3585. [Google Scholar] [CrossRef]
- Rutar, M.; Natoli, R.; Provis, J.M. Small interfering RNA-mediated suppression of Ccl2 in Muller cells attenuates microglial recruitment and photoreceptor death following retinal degeneration. J. Neuroinflamm. 2012, 9, 221. [Google Scholar] [CrossRef]
- Suzuki, M.; Tsujikawa, M.; Itabe, H.; Du, Z.J.; Xie, P.; Matsumura, N.; Fu, X.; Zhang, R.; Sonoda, K.H.; Egashira, K.; et al. Chronic photo-oxidative stress and subsequent MCP-1 activation as causative factors for age-related macular degeneration. J. Cell Sci. 2012, 125 Pt 10, 2407–2415. [Google Scholar] [CrossRef] [Green Version]
- Kohno, H.; Chen, Y.; Kevany, B.M.; Pearlman, E.; Miyagi, M.; Maeda, T.; Palczewski, K.; Maeda, A. Photoreceptor Proteins Initiate Microglial Activation via Toll-like Receptor 4 in Retinal Degeneration Mediated by All-trans-retinal. J. Biol. Chem. 2013, 288, 15326–15341. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Guilloty, F.; Saeed, A.M.; Echegaray, J.J.; Duffort, S.; Ballmick, A.; Tan, Y.; Betancourt, M.; Viteri, E.; Ramkhellawan, G.C.; Ewald, E.; et al. Infiltration of proinflammatory m1 macrophages into the outer retina precedes damage in a mouse model of age-related macular degeneration. Int. J. Inflam. 2013, 2013, 503725. [Google Scholar] [CrossRef]
- Hu, S.J.; Calippe, B.; Lavalette, S.; Roubeix, C.; Montassar, F.; Housset, M.; Levy, O.; Delarasse, C.; Paques, M.; Sahel, J.A.; et al. Upregulation of P2RX7 in Cx3cr1-Deficient Mononuclear Phagocytes Leads to Increased Interleukin-1beta Secretion and Photoreceptor Neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 6987–6996. [Google Scholar] [CrossRef]
- Lavalette, S.; Raoul, W.; Houssier, M.; Camelo, S.; Levy, O.; Calippe, B.; Jonet, L.; Behar-Cohen, F.; Chemtob, S.; Guillonneau, X.; et al. Interleukin-1beta inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration. Am. J. Pathol. 2011, 178, 2416–2423. [Google Scholar] [CrossRef] [PubMed]
- Mathis, T.; Housset, M.; Eandi, C.; Beguier, F.; Touhami, S.; Reichman, S.; Augustin, S.; Gondouin, P.; Sahel, J.A.; Kodjikian, L.; et al. Activated monocytes resist elimination by retinal pigment epithelium and downregulate their OTX2 expression via TNF-alpha. Aging Cell 2017, 16, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Izumi-Nagai, K.; Nagai, N.; Ozawa, Y.; Mihara, M.; Ohsugi, Y.; Kurihara, T.; Koto, T.; Satofuka, S.; Inoue, M.; Tsubota, K.; et al. Interleukin-6 receptor-mediated activation of signal transducer and activator of transcription-3 (STAT3) promotes choroidal neovascularization. Am. J. Pathol. 2007, 170, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Vitiello, L.; Torcinaro, A.; Ferraro, E. The Role of Metabolic Remodeling in Macrophage Polarization and Its Effect on Skeletal Muscle Regeneration. Antiox. Redox Signal. 2018. [Google Scholar] [CrossRef]
- El Kasmi, K.C.; Stenmark, K.R. Contribution of metabolic reprogramming to macrophage plasticity and function. Semin. Immunol. 2015, 27, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvan-Pena, S.; O’Neill, L.A. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar]
- Rath, M.; Muller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef]
- Kobzik, L. Translating NO biology into clinical advances: Still searching for the right dictionary? Am. J. Respir. Cell Mol. Biol. 2009, 41, 9–13. [Google Scholar] [CrossRef]
- St Clair, E.W.; Wilkinson, W.E.; Lang, T.; Sanders, L.; Misukonis, M.A.; Gilkeson, G.S.; Pisetsky, D.S.; Granger, D.I.; Weinberg, J.B. Increased expression of blood mononuclear cell nitric oxide synthase type 2 in rheumatoid arthritis patients. J. Exp. Med. 1996, 184, 1173–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherepanoff, S.; McMenamin, P.; Gillies, M.C.; Kettle, E.; Sarks, S.H. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br. J. Ophthalmol. 2010, 94, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Doulias, P.T.; Tenopoulou, M.; Greene, J.L.; Raju, K.; Ischiropoulos, H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal. 2013, 6, rs1. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Song, H.; Yin, L.; Rizzo, M.G.; Sidhu, R.; Covey, D.F.; Ory, D.S.; Semenkovich, C.F. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature 2016, 539, 294–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumann, J. It is all about fluidity: Fatty acids and macrophage phagocytosis. Eur. J. Pharmacol. 2016, 785, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [Green Version]
- McFadden, B.A.; Purohit, S. Itaconate, an isocitrate lyase-directed inhibitor in Pseudomonas indigofera. J. Bacteriol. 1977, 131, 136–144. [Google Scholar]
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem. 2016, 291, 14274–14284. [Google Scholar] [CrossRef] [Green Version]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, 580–585. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Miao, F.J.; Lin, D.C.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapieha, P.; Sirinyan, M.; Hamel, D.; Zaniolo, K.; Joyal, J.S.; Cho, J.H.; Honore, J.C.; Kermorvant-Duchemin, E.; Varma, D.R.; Tremblay, S.; et al. The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat. Med. 2008, 14, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Bisetto, S.; Whitaker-Menezes, D.; Wilski, N.A.; Tuluc, M.; Curry, J.; Zhan, T.; Snyder, C.M.; Martinez-Outschoorn, U.E.; Philp, N.J. Monocarboxylate Transporter 4 (MCT4) Knockout Mice Have Attenuated 4NQO Induced Carcinogenesis; A Role for MCT4 in Driving Oral Squamous Cell Cancer. Front. Oncol. 2018, 8, 324. [Google Scholar] [CrossRef]
- Tan, Z.; Xie, N.; Banerjee, S.; Cui, H.; Fu, M.; Thannickal, V.J.; Liu, G. The monocarboxylate transporter 4 is required for glycolytic reprogramming and inflammatory response in macrophages. J. Biol. Chem. 2015, 290, 46–55. [Google Scholar] [CrossRef]
- Spolarics, Z.; Navarro, L. Endotoxin stimulates the expression of glucose-6-phosphate dehydrogenase in Kupffer and hepatic endothelial cells. J. Leukoc. Biol. 1994, 56, 453–457. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mameza, M.G.; Lee, Y.S.; Eseonu, C.I.; Yu, C.R.; Kang Derwent, J.J.; Egwuagu, C.E. Suppressors of cytokine-signaling proteins induce insulin resistance in the retina and promote survival of retinal cells. Diabetes 2008, 57, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, R.; Zhang, S.; Wei, X.; Doggett, T.; Adak, S.; Enright, J.; Shah, V.; Ling, G.; Chen, S.; Yoshino, J.; et al. Retinal de novo lipogenesis coordinates neurotrophic signaling to maintain vision. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antiox. Redox Signal. 2017, 26, 462–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef]
- McKay, G.J.; Patterson, C.C.; Chakravarthy, U.; Dasari, S.; Klaver, C.C.; Vingerling, J.R.; Ho, L.; de Jong, P.T.; Fletcher, A.E.; Young, I.S.; et al. Evidence of association of APOE with age-related macular degeneration: A pooled analysis of 15 studies. Hum. Mutat. 2011, 32, 1407–1416. [Google Scholar] [CrossRef]
- Levy, O.; Lavalette, S.; Hu, S.J.; Housset, M.; Raoul, W.; Eandi, C.; Sahel, J.A.; Sullivan, P.M.; Guillonneau, X.; Sennlaub, F. APOE-isoforms control pathogenic subretinal inflammation in age related macular degeneration. J. Neurosci. 2015, 35, 13568–13576. [Google Scholar] [CrossRef]
- Calippe, B.; Guillonneau, X.; Sennlaub, F. Complement factor H and related proteins in age-related macular degeneration. Comptes Rendus Biologies 2014, 337, 178–184. [Google Scholar] [CrossRef]
- Philp, N.J.; Yoon, H.; Grollman, E.F. Monocarboxylate transporter MCT1 is located in the apical membrane and MCT3 in the basal membrane of rat RPE. Am. J. Physiol. 1998, 274 Pt 2, R1824–R1828. [Google Scholar] [CrossRef]
- Leveillard, T.; Sahel, J.A. Metabolic and redox signaling in the retina. Cell Mol. Life Sci. 2017, 74, 3649–3665. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Vogt, S.D.; Curcio, C.A.; Wang, L.; Li, C.M.; McGwin, G., Jr.; Medeiros, N.E.; Philp, N.J.; Kimble, J.A.; Read, R.W. Retinal pigment epithelial expression of complement regulator CD46 is altered early in the course of geographic atrophy. Exp. Eye Res. 2011, 93, 413–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Fritsche, L.G.; Zhou, X.; Abecasis, G. International Age-Related Macular Degeneration Genomics, C. A Scalable Bayesian Method for Integrating Functional Information in Genome-wide Association Studies. Am. J. Hum. Genet. 2017, 101, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Monden, I.; Olsowski, A.; Krause, G.; Keller, K. The large cytoplasmic loop of the glucose transporter GLUT1 is an essential structural element for function. Biol. Chem. 2001, 382, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Flatt, J.F.; Guizouarn, H.; Burton, N.M.; Borgese, F.; Tomlinson, R.J.; Forsyth, R.J.; Baldwin, S.A.; Levinson, B.E.; Quittet, P.; Aguilar-Martinez, P.; et al. Stomatin-deficient cryohydrocytosis results from mutations in SLC2A1: A novel form of GLUT1 deficiency syndrome. Blood 2011, 118, 5267–5277. [Google Scholar] [CrossRef]
- Leveillard, T.; Ait-Ali, N. Cell Signaling with Extracellular Thioredoxin and Thioredoxin-Like Proteins: Insight into Their Mechanisms of Action. Oxid. Med. Cell Longev. 2017, 2017, 8475125. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Léveillard, T.; Philp, N.J.; Sennlaub, F. Is Retinal Metabolic Dysfunction at the Center of the Pathogenesis of Age-related Macular Degeneration? Int. J. Mol. Sci. 2019, 20, 762. https://doi.org/10.3390/ijms20030762
Léveillard T, Philp NJ, Sennlaub F. Is Retinal Metabolic Dysfunction at the Center of the Pathogenesis of Age-related Macular Degeneration? International Journal of Molecular Sciences. 2019; 20(3):762. https://doi.org/10.3390/ijms20030762
Chicago/Turabian StyleLéveillard, Thierry, Nancy J. Philp, and Florian Sennlaub. 2019. "Is Retinal Metabolic Dysfunction at the Center of the Pathogenesis of Age-related Macular Degeneration?" International Journal of Molecular Sciences 20, no. 3: 762. https://doi.org/10.3390/ijms20030762
APA StyleLéveillard, T., Philp, N. J., & Sennlaub, F. (2019). Is Retinal Metabolic Dysfunction at the Center of the Pathogenesis of Age-related Macular Degeneration? International Journal of Molecular Sciences, 20(3), 762. https://doi.org/10.3390/ijms20030762