Copy-Paste Mutagenesis: A Method for Large-Scale Alteration of Viral Genomes



Abstract
:1. Introduction
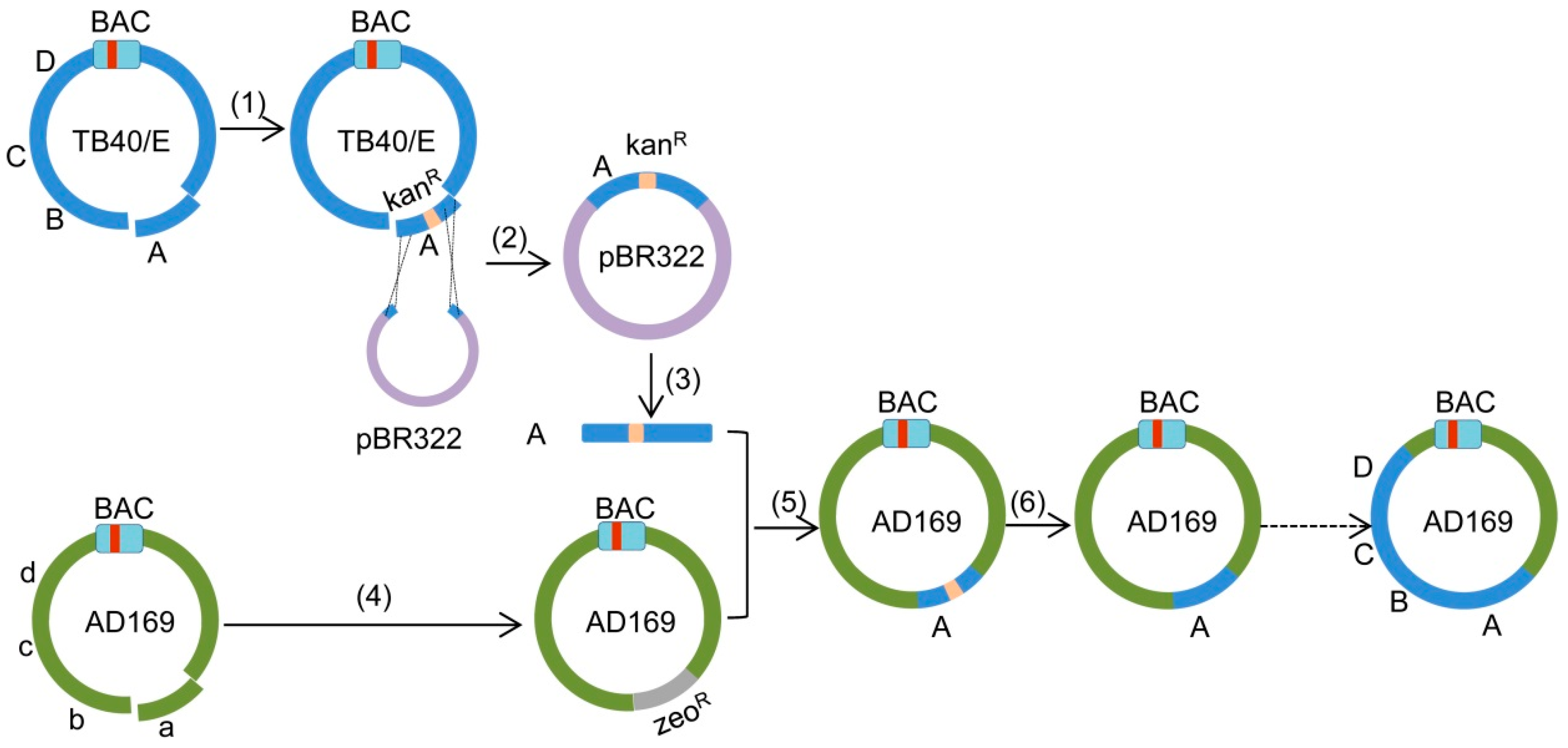
2. Results
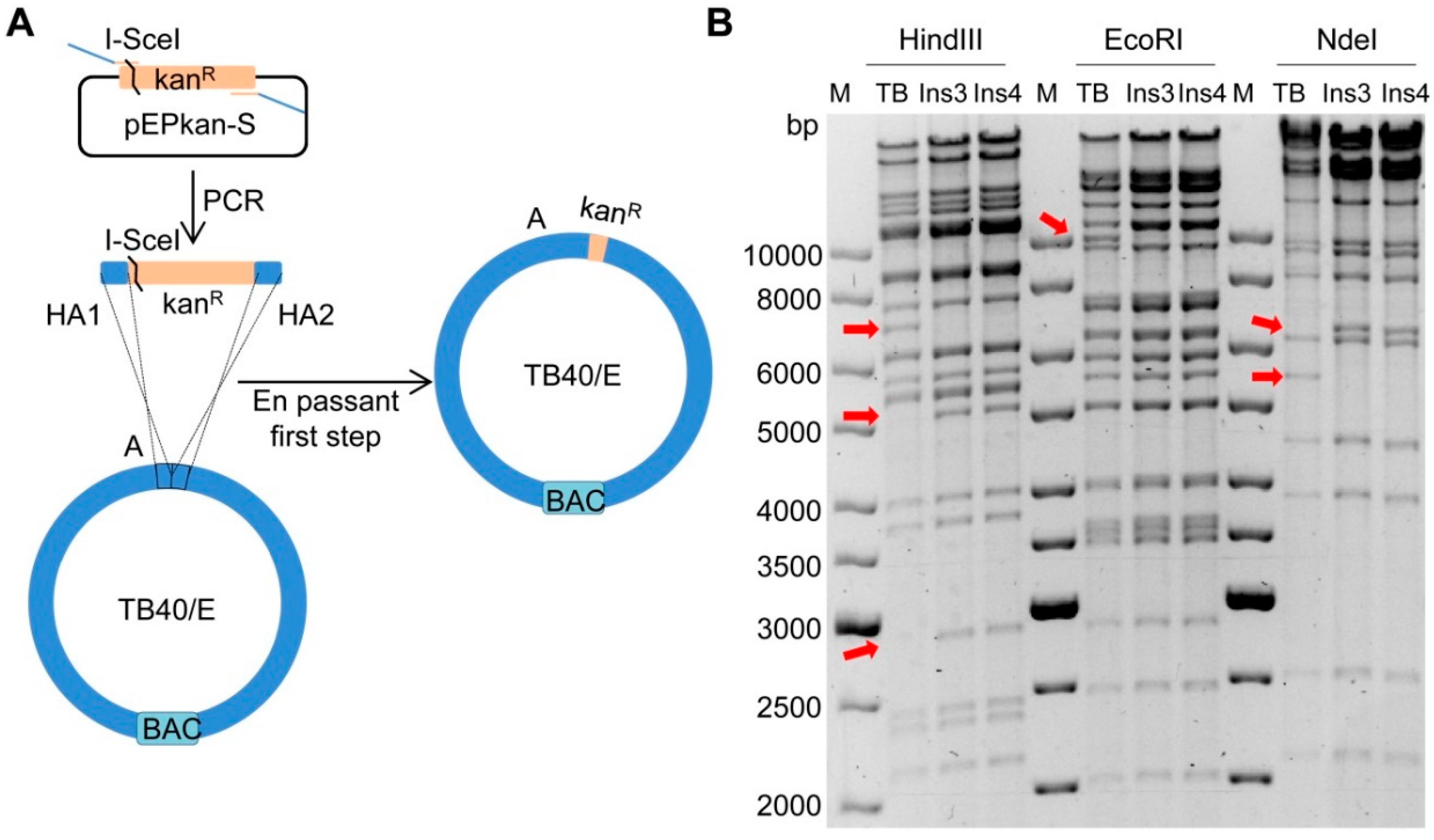
2.1. Insertion of a Selectable Marker into the TB40/E Target Fragment
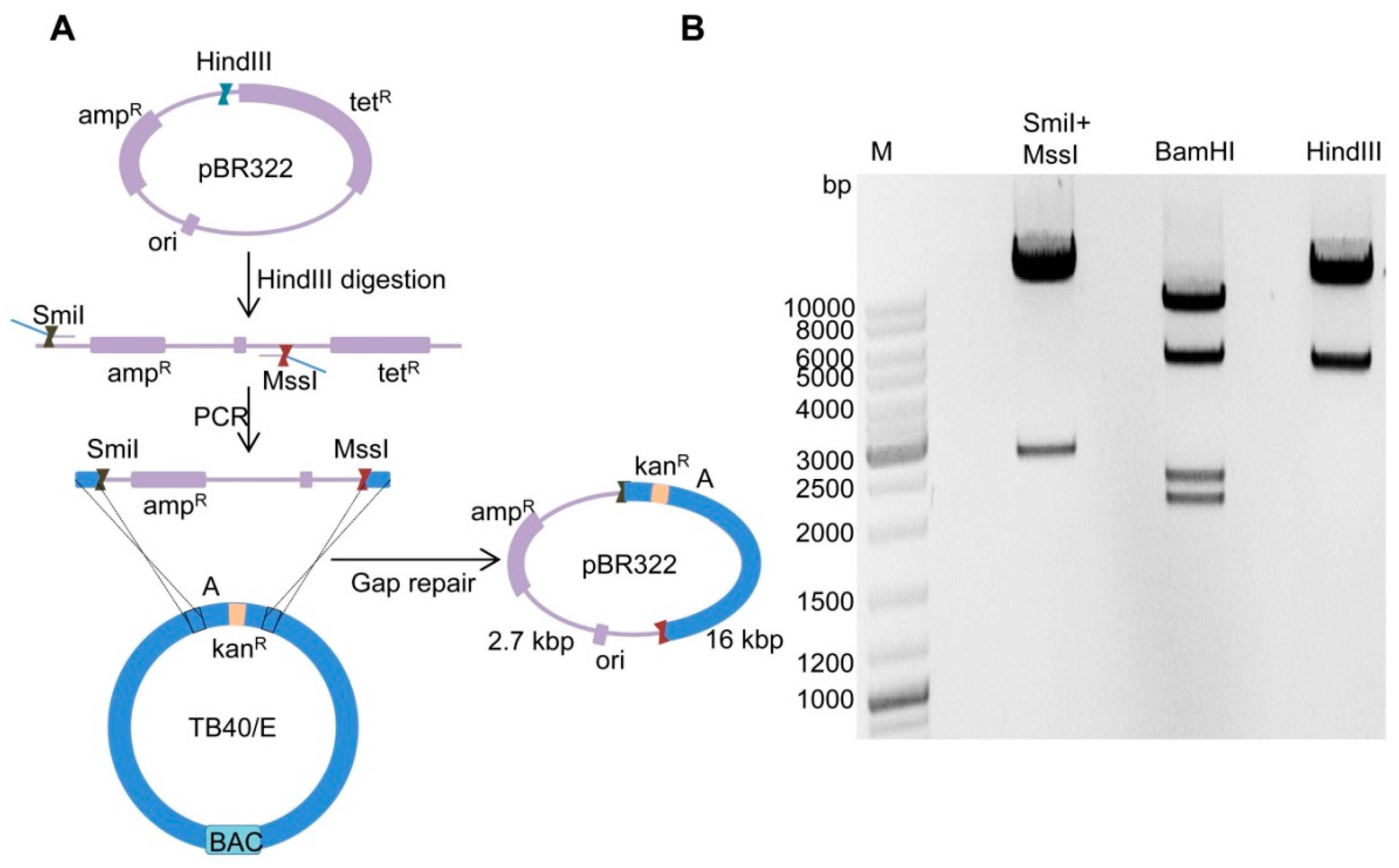
2.2. Cloning of Fragment ‘A-Ins’ in Plasmid pBR322
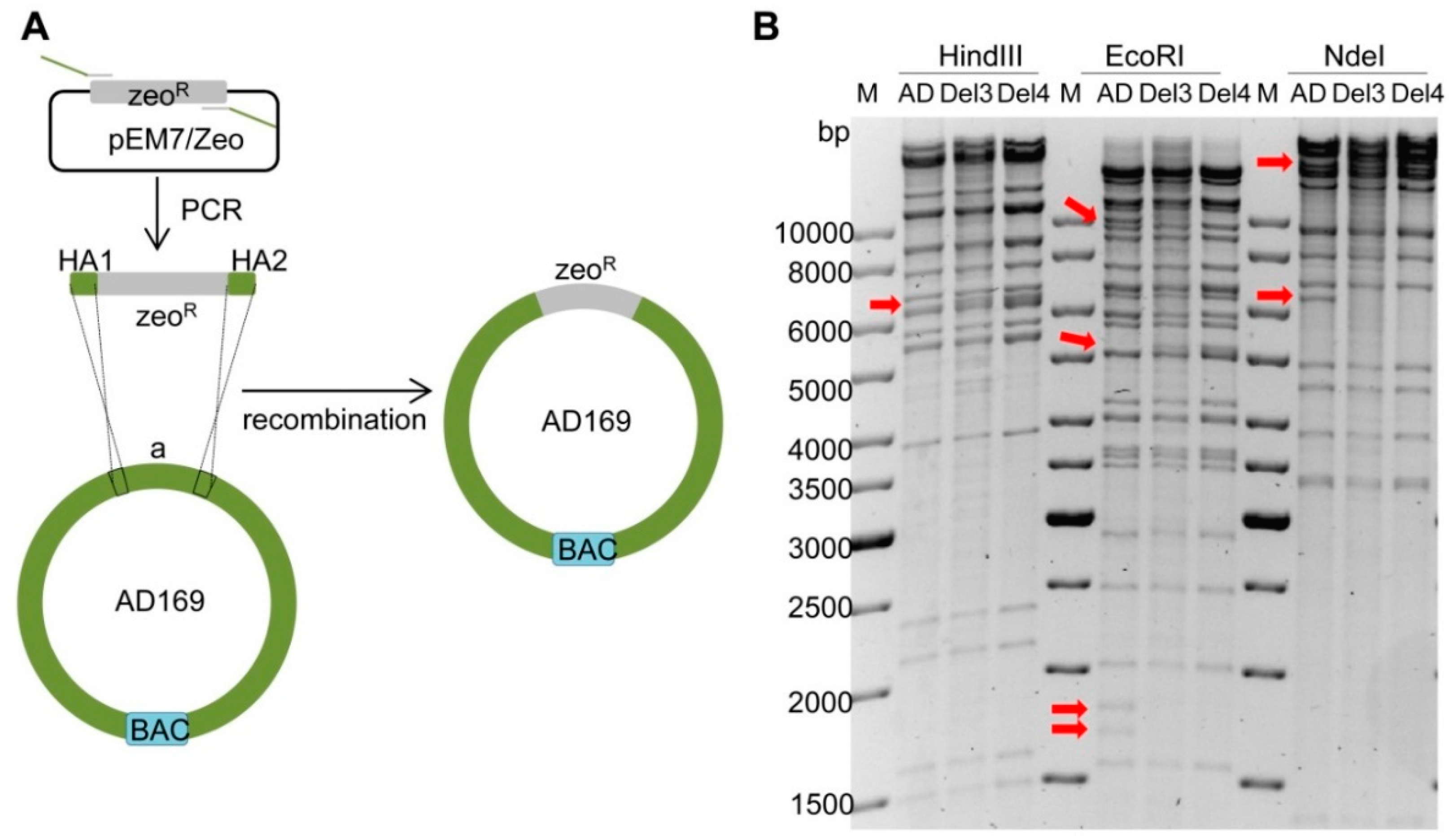
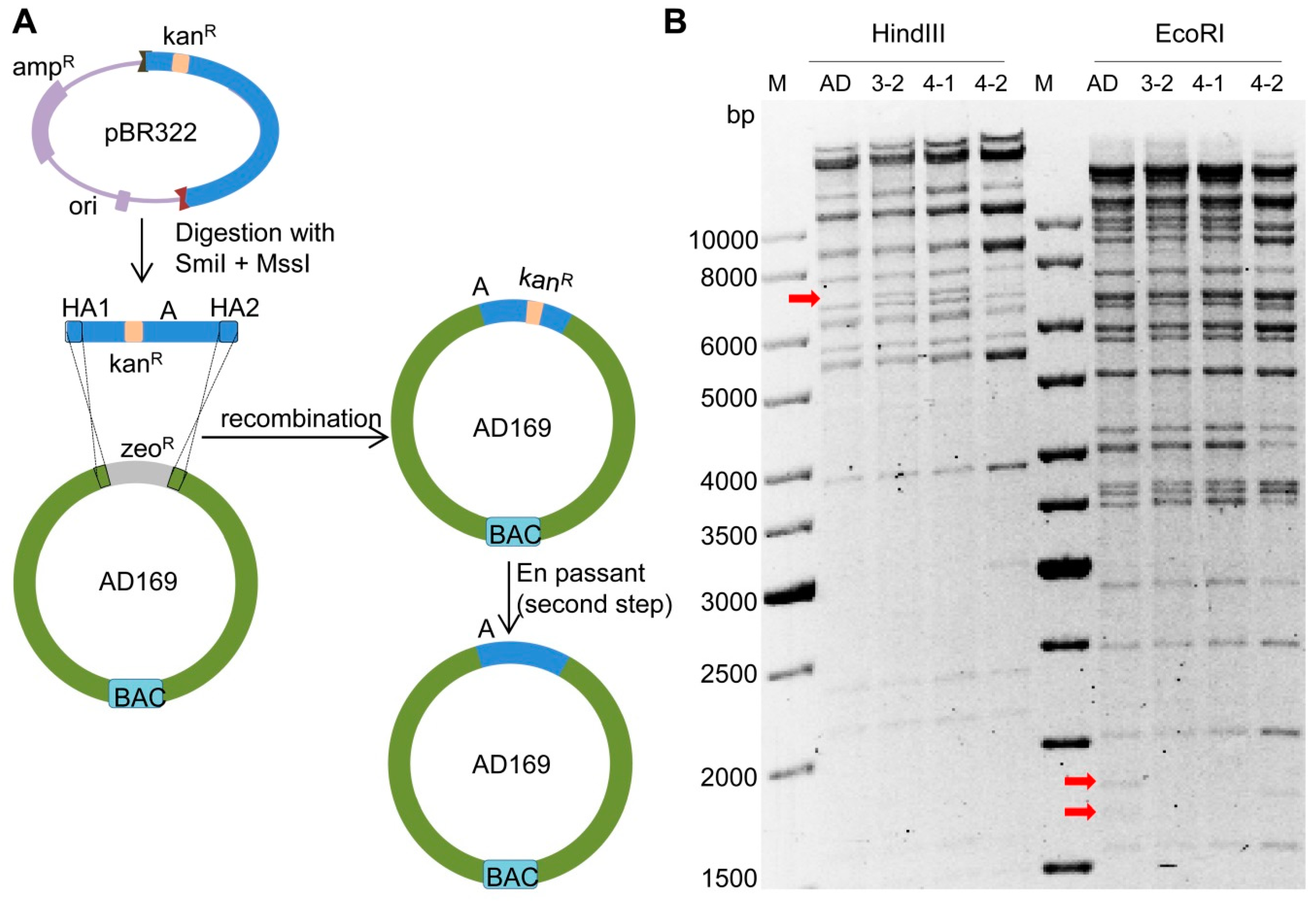
2.3. Deletion of Fragment ‘a’ in AD169
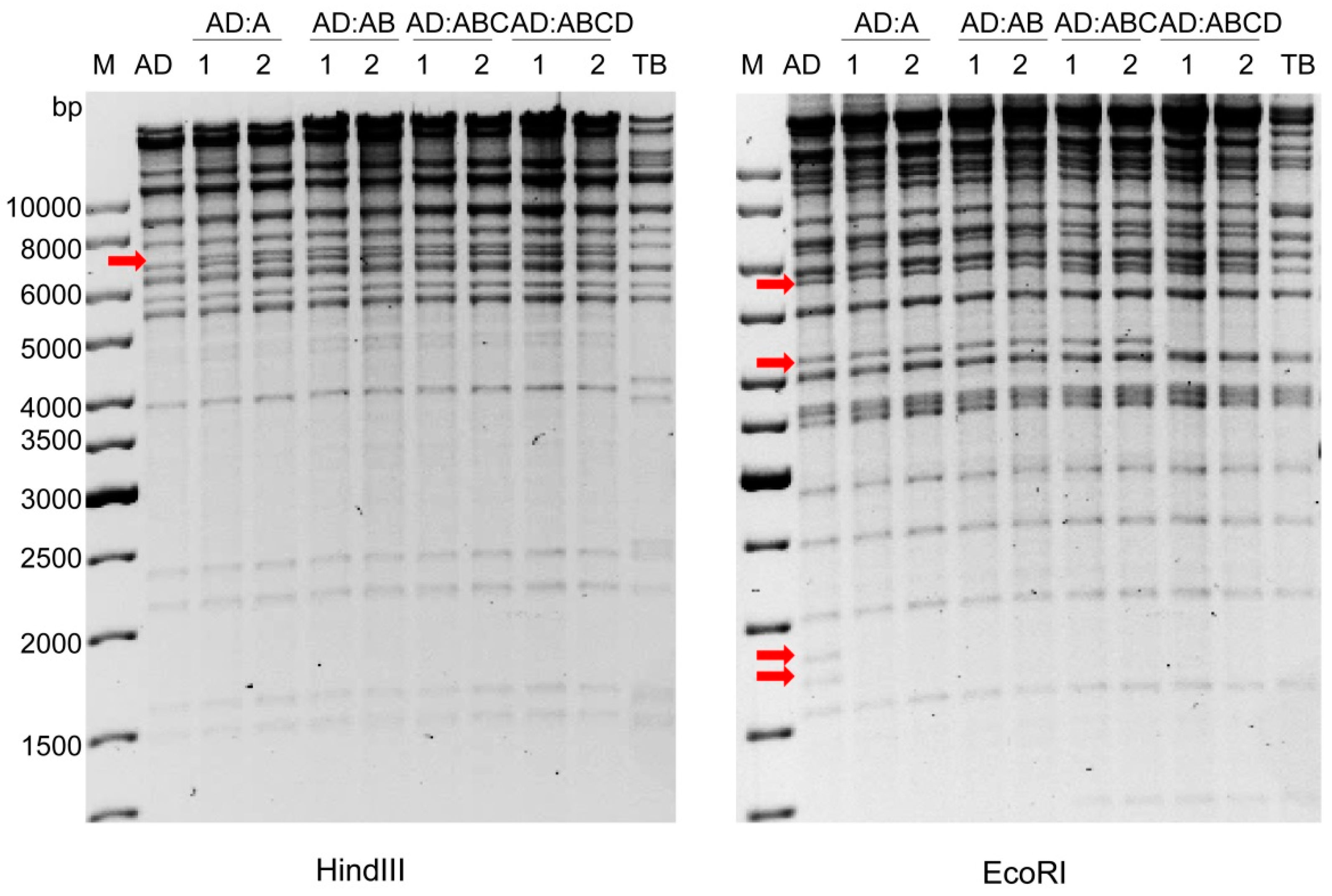
2.4. Insertion of Fragment ‘A-Ins’ into AD169-a-Del
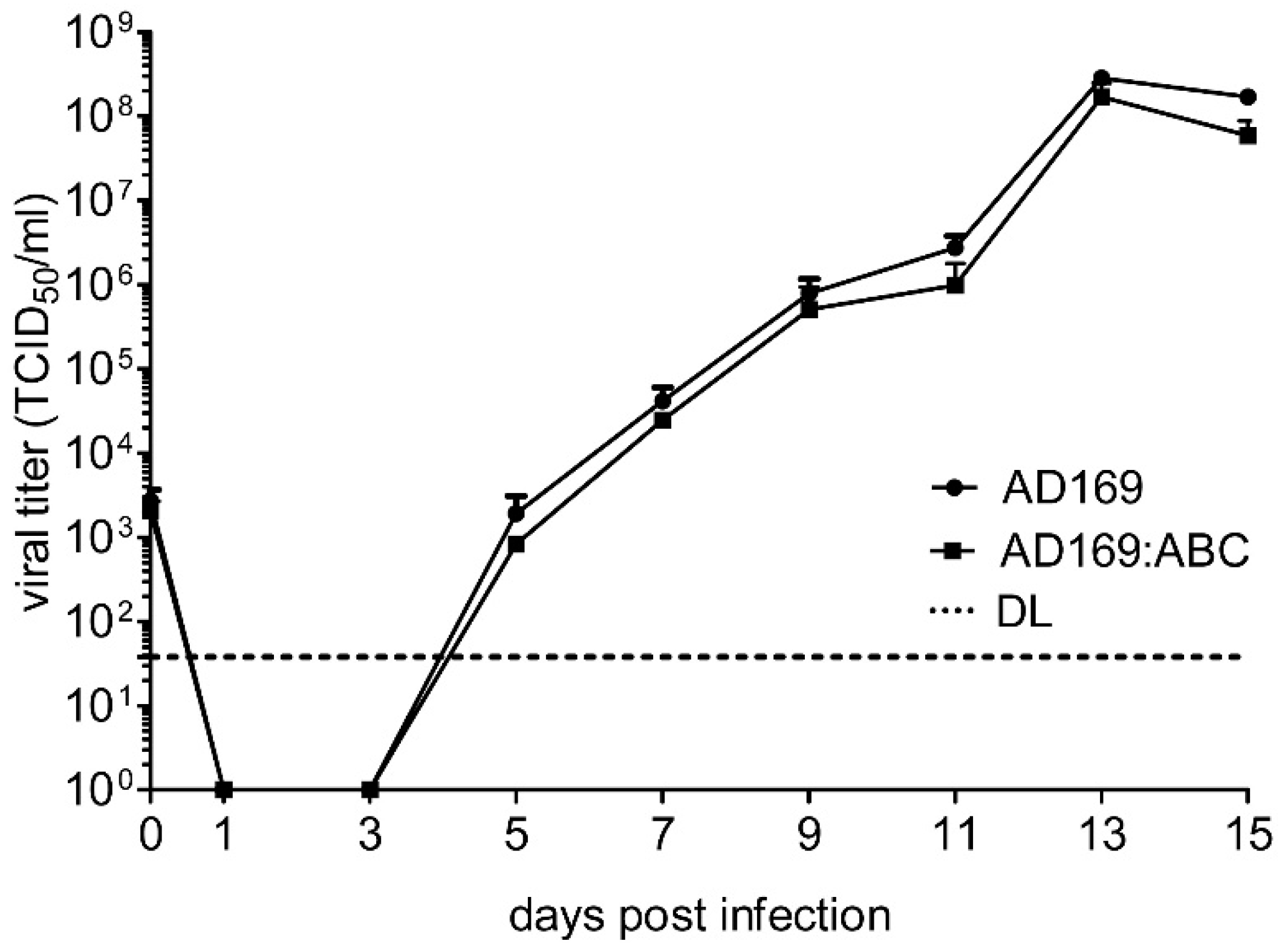
2.5. Reconstitution of Chimeric Virus from BAC DNA
3. Discussion
4. Materials and Methods
4.1. Plasmids and Reagents
4.2. Cells and Viruses
4.3. BAC Mutagenesis
4.4. Gap Repair Cloning
4.5. BAC Sequence Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HCMV | Human cytomegalovirus |
| BAC | Bacterial artificial chromosome |
| HA | Homology arm |
References
- Pellet, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1802–18022. [Google Scholar]
- Mocarski, E.S.; Shenk, T.; Griffiths, P.D.; Pass, R.F. Cytomegaloviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1960–2014. [Google Scholar]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Yu, D.; Grimwood, J.; Schmutz, J.; Dickson, M.; Jarvis, M.A.; Hahn, G.; Nelson, J.A.; Myers, R.M.; Shenk, T.E. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2003, 100, 14976–14981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C.; et al. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern-Ginossar, N.; Weisburd, B.; Michalski, A.; Le, V.T.; Hein, M.Y.; Huang, S.X.; Ma, M.; Shen, B.; Qian, S.B.; Hengel, H.; et al. Decoding human cytomegalovirus. Science 2012, 338, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Erhard, F.; Halenius, A.; Zimmermann, C.; L’Hernault, A.; Kowalewski, D.J.; Weekes, M.P.; Stevanovic, S.; Zimmer, R.; Dölken, L. Improved Ribo-seq enables identification of cryptic translation events. Nat. Methods 2018, 15, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Greaves, R.F.; Brown, J.M.; Vieira, J.; Mocarski, E.S. Selectable insertion and deletion mutagenesis of the human cytomegalovirus genome using the Escherichia coli guanosine phosphoribosyl transferase (gpt) gene. J. Gen. Virol. 1995, 76 Pt 9, 2151–2160. [Google Scholar] [CrossRef]
- Spaete, R.R.; Mocarski, E.S. Insertion and deletion mutagenesis of the human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 1987, 84, 7213–7217. [Google Scholar] [CrossRef] [PubMed]
- Messerle, M.; Crnkovic, I.; Hammerschmidt, W.; Ziegler, H.; Koszinowski, U.H. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 1997, 94, 14759–14763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, E.M.; Hahn, G.; Koszinowski, U.H.; Messerle, M. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: A new approach for construction of HCMV mutants. J. Virol. 1999, 73, 8320–8329. [Google Scholar] [PubMed]
- Yu, D.; Smith, G.A.; Enquist, L.W.; Shenk, T. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 2002, 76, 2316–2328. [Google Scholar] [CrossRef] [PubMed]
- Brune, W.; Messerle, M.; Koszinowski, U.H. Forward with BACs: New tools for herpesvirus genomics. Trends Genet. 2000, 16, 254–259. [Google Scholar] [CrossRef]
- Brune, W.; Wagner, M.; Messerle, M. Manipulating cytomegalovirus genomes by BAC mutagenesis: Strategies and applications. In Cytomegaloviruses: Molecular Biology and Immunology; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2006; pp. 61–89. [Google Scholar]
- Warden, C.; Tang, Q.; Zhu, H. Herpesvirus BACs: Past, present, and future. J. Biomed. Biotechnol. 2011, 2011, 124595. [Google Scholar] [CrossRef] [PubMed]
- Cottingham, M.G. Genetic manipulation of poxviruses using bacterial artificial chromosome recombineering. Methods Mol. Biol. 2012, 890, 37–57. [Google Scholar] [PubMed]
- Roy, P.; Noad, R. Use of bacterial artificial chromosomes in baculovirus research and recombinant protein expression: Current trends and future perspectives. ISRN Microbiol. 2012, 2012, 628797. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.N.; Meers, J.; Fowler, E.; Mahony, T. Back to BAC: The use of infectious clone technologies for viral mutagenesis. Viruses 2012, 4, 211–235. [Google Scholar] [CrossRef] [PubMed]
- Brune, W.; Ménard, C.; Hobom, U.; Odenbreit, S.; Messerle, M.; Koszinowski, U.H. Rapid identification of essential and nonessential herpesvirus genes by direct transposon mutagenesis. Nat. Biotechnol. 1999, 17, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Enquist, L.W. Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J. Virol. 1999, 73, 6405–6414. [Google Scholar]
- Brune, W. Random transposon mutagenesis of large DNA molecules in Escherichia coli. Methods Mol. Biol. 2002, 182, 165–171. [Google Scholar]
- Yu, D.; Silva, M.C.; Shenk, T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 12396–12401. [Google Scholar] [CrossRef] [Green Version]
- Muyrers, J.P.; Zhang, Y.; Testa, G.; Stewart, A.F. Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res. 1999, 27, 1555–1557. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Ellis, H.M.; Lee, E.C.; Jenkins, N.A.; Copeland, N.G.; Court, D.L. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 5978–5983. [Google Scholar] [CrossRef]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warming, S.; Costantino, N.; Court, D.L.; Jenkins, N.A.; Copeland, N.G. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005, 33, e36. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [PubMed]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En passant mutagenesis: A two step markerless red recombination system. Methods Mol. Biol. 2010, 634, 421–430. [Google Scholar] [PubMed]
- Lee, E.C.; Yu, D.; Martinez de Velasco, J.; Tessarollo, L.; Swing, D.A.; Court, D.L.; Jenkins, N.A.; Copeland, N.G. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 2001, 73, 56–65. [Google Scholar] [CrossRef]
- Renzette, N.; Bhattacharjee, B.; Jensen, J.D.; Gibson, L.; Kowalik, T.F. Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog. 2011, 7, e1001344. [Google Scholar] [CrossRef]
- Heineman, T.C.; Schleiss, M.; Bernstein, D.I.; Spaete, R.R.; Yan, L.; Duke, G.; Prichard, M.; Wang, Z.; Yan, Q.; Sharp, M.A.; et al. A phase 1 study of 4 live, recombinant human cytomegalovirus Towne/Toledo chimeric vaccines. J. Infect. Dis. 2006, 193, 1350–1360. [Google Scholar] [CrossRef]
- Cha, T.A.; Tom, E.; Kemble, G.W.; Duke, G.M.; Mocarski, E.S.; Spaete, R.R. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 1996, 70, 78–83. [Google Scholar]
- Bradley, A.J.; Lurain, N.S.; Ghazal, P.; Trivedi, U.; Cunningham, C.; Baluchova, K.; Gatherer, D.; Wilkinson, G.W.; Dargan, D.J.; Davison, A.J. High-throughput sequence analysis of variants of human cytomegalovirus strains Towne and AD169. J. Gen. Virol. 2009, 90, 2375–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Shenk, T. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J. Virol. 2005, 79, 10330–10338. [Google Scholar] [CrossRef] [PubMed]
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; et al. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 2004, 78, 10023–10033. [Google Scholar] [CrossRef] [PubMed]
- Skaletskaya, A.; Bartle, L.M.; Chittenden, T.; McCormick, A.L.; Mocarski, E.S.; Goldmacher, V.S. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. USA 2001, 98, 7829–7834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchini, A.; Liu, H.; Zhu, H. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol. 2001, 75, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Hahn, G.; Rose, D.; Wagner, M.; Rhiel, S.; McVoy, M.A. Cloning of the genomes of human cytomegalovirus strains Toledo, TownevarRIT3, and Towne long as BACs and site-directed mutagenesis using a PCR-based technique. Virology 2003, 307, 164–177. [Google Scholar] [CrossRef]
- Sinzger, C.; Hahn, G.; Digel, M.; Katona, R.; Sampaio, K.L.; Messerle, M.; Hengel, H.; Koszinowski, U.; Brune, W.; Adler, B. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 2008, 89, 359–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanton, R.J.; Baluchova, K.; Dargan, D.J.; Cunningham, C.; Sheehy, O.; Seirafian, S.; McSharry, B.P.; Neale, M.L.; Davies, J.A.; Tomasec, P.; et al. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J. Clin. Investig. 2010, 120, 3191–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemble, G.; Duke, G.; Winter, R.; Spaete, R. Defined large-scale alterations of the human cytomegalovirus genome constructed by cotransfection of overlapping cosmids. J. Virol. 1996, 70, 2044–2048. [Google Scholar] [PubMed]
- Rivero-Muller, A.; Lajic, S.; Huhtaniemi, I. Assisted large fragment insertion by Red/ET-recombination (ALFIRE)—An alternative and enhanced method for large fragment recombineering. Nucleic Acids Res. 2007, 35, e78. [Google Scholar] [CrossRef] [PubMed]
- Kuhlman, T.E.; Cox, E.C. Site-specific chromosomal integration of large synthetic constructs. Nucleic Acids Res. 2010, 38, e92. [Google Scholar] [CrossRef] [PubMed]
- Baldick, C.J., Jr.; Marchini, A.; Patterson, C.E.; Shenk, T. Human cytomegalovirus tegument protein pp71 (ppUL82) enhances the infectivity of viral DNA and accelerates the infectious cycle. J. Virol. 1997, 71, 4400–4408. [Google Scholar] [PubMed]
- Le, V.T.; Trilling, M.; Hengel, H. The cytomegaloviral protein pUL138 acts as potentiator of tumor necrosis factor (TNF) receptor 1 surface density to enhance ULb’-encoded modulation of TNF-alpha signaling. J. Virol. 2011, 85, 13260–13270. [Google Scholar] [CrossRef]
- Mahy, B.W.J.; Kangro, H.O. Virology Methods Manual; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Brune, W.; Nevels, M.; Shenk, T. Murine cytomegalovirus m41 open reading frame encodes a Golgi-localized antiapoptotic protein. J. Virol. 2003, 77, 11633–11643. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, E.; Spohn, M.; Indenbirken, D.; Brune, W. Complete Genome Sequence of a Human Cytomegalovirus Strain AD169 Bacterial Artificial Chromosome Clone. Genome Announc. 2016, 4, e00091-16. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequences * | Purpose |
|---|---|---|
| A-MssI-Fwd | CCCCGTAAACGATATAAGCGCTATCGCCAGATATCGCGTA gtttaaacGATACGCGAGCGAACGTGA | Cloning of UL112-127 of TB40/E in pBR322 |
| A-SmiI-Rev | AAACTACGTCACCCGACACGCGGAAAAGAAAGACCGTCGC atttaaatTTCTTAGACGTCAGGTGGCAC | |
| a-Del-Fwd | TCCTCTTGTAGCAACGTGAGGACGACTACTCCGTGTGGCTCGACGGTACGTGTTGACAATTAATCATCGGCAT | Deletion of UL112-127 in AD169 and replacement with zeoR |
| a-Del-Rev | GTGTGTCGCAAATATCGCAGTTTCGATATAGGTGACAGACGATATGAGGCTCAGTCCTGCTCCTCGGCCA | |
| A-Ins-Fwd | CCATTTACCGTAAGTTATGTAACGCGGAACTCCATATATGGGCTATGAACTAATGACCCCTAGGGATAACAGGGTAATCGATTT | Insertion of kanR/I-SceI cassette into UL112-127 of TB40/E |
| A-Ins-Rev | GACATTGATTATTGACTAGTTATTAATAGTAATCAATTACGGGGTCATTAGTTCATAGCCGCCAGTGTTACAACCAATTAACC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.; Brixel, R.; Brune, W. Copy-Paste Mutagenesis: A Method for Large-Scale Alteration of Viral Genomes. Int. J. Mol. Sci. 2019, 20, 913. https://doi.org/10.3390/ijms20040913
Tang J, Brixel R, Brune W. Copy-Paste Mutagenesis: A Method for Large-Scale Alteration of Viral Genomes. International Journal of Molecular Sciences. 2019; 20(4):913. https://doi.org/10.3390/ijms20040913
Chicago/Turabian StyleTang, Jiajia, Renke Brixel, and Wolfram Brune. 2019. "Copy-Paste Mutagenesis: A Method for Large-Scale Alteration of Viral Genomes" International Journal of Molecular Sciences 20, no. 4: 913. https://doi.org/10.3390/ijms20040913
APA StyleTang, J., Brixel, R., & Brune, W. (2019). Copy-Paste Mutagenesis: A Method for Large-Scale Alteration of Viral Genomes. International Journal of Molecular Sciences, 20(4), 913. https://doi.org/10.3390/ijms20040913