Hydroxamic Acid-Based Histone Deacetylase (HDAC) Inhibitors Bearing a Pyrazole Scaffold and a Cinnamoyl Linker


 ,
, 
 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
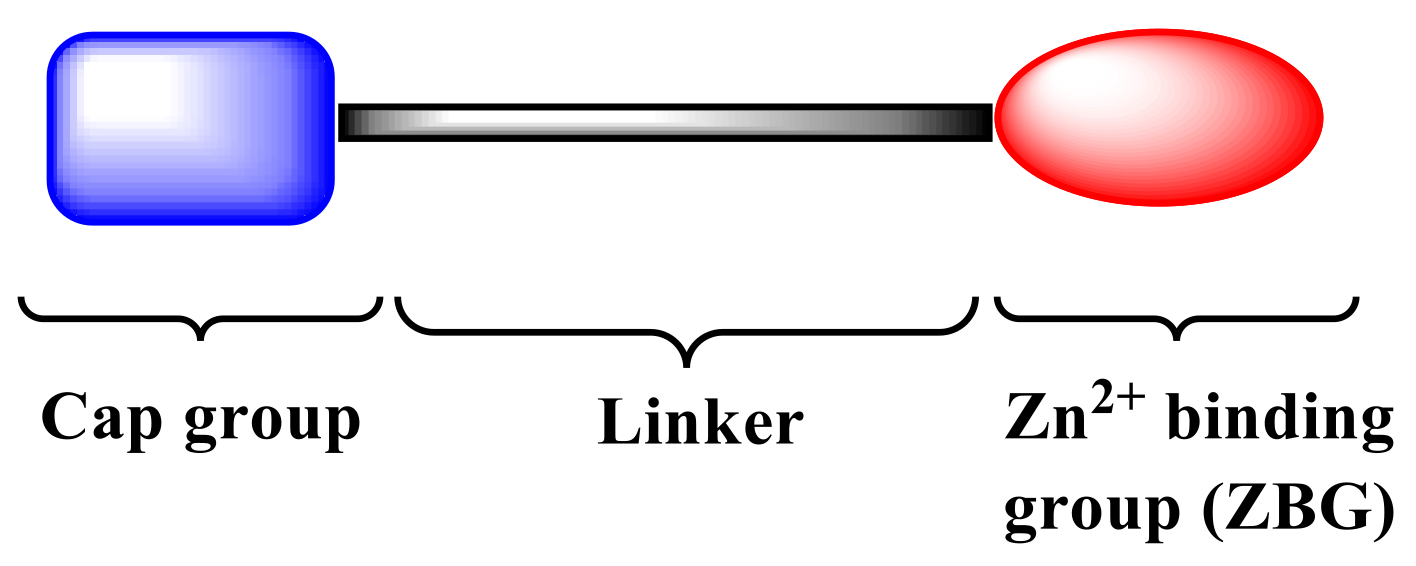
:1. Introduction
2. Results and Discussion
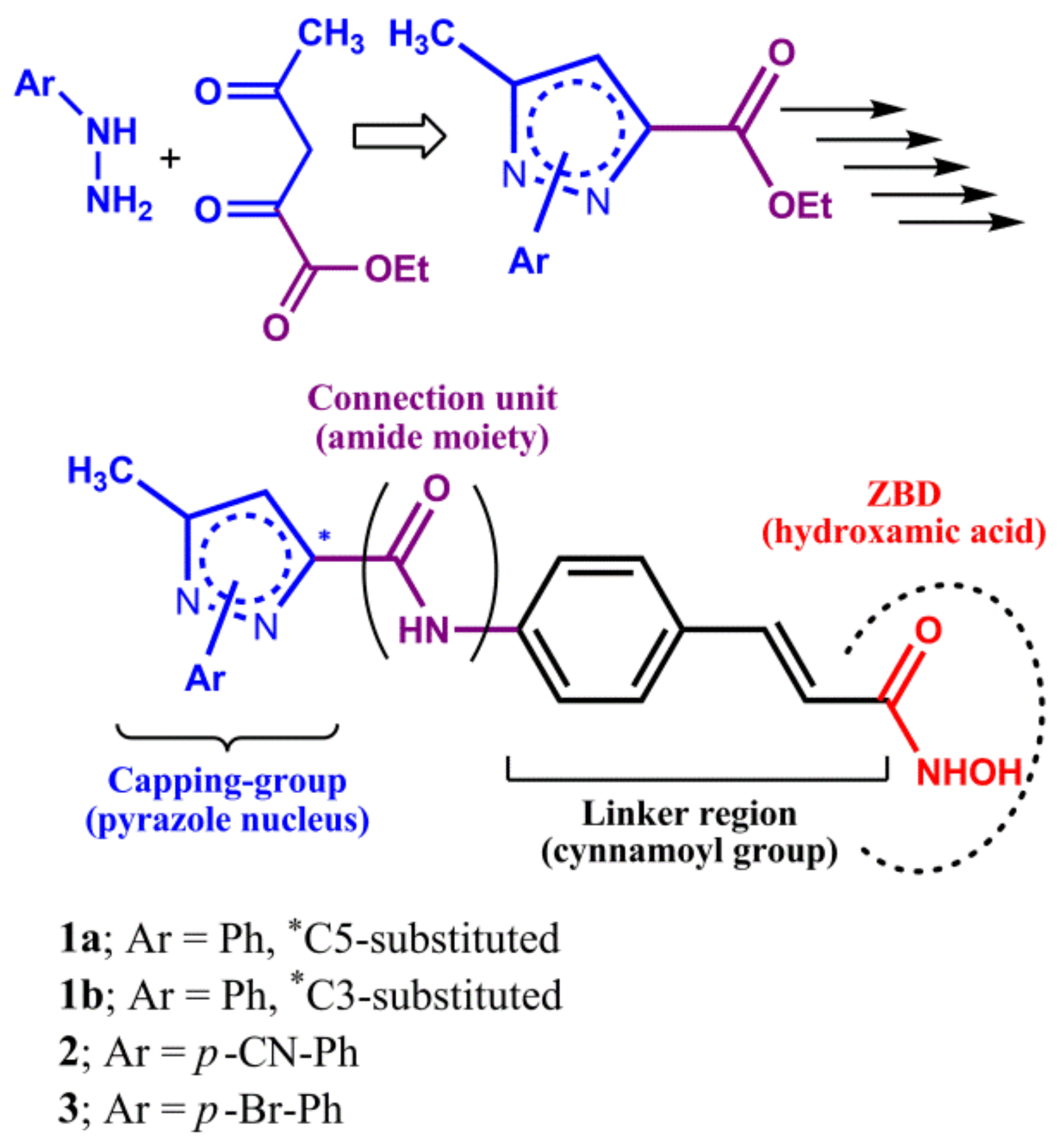
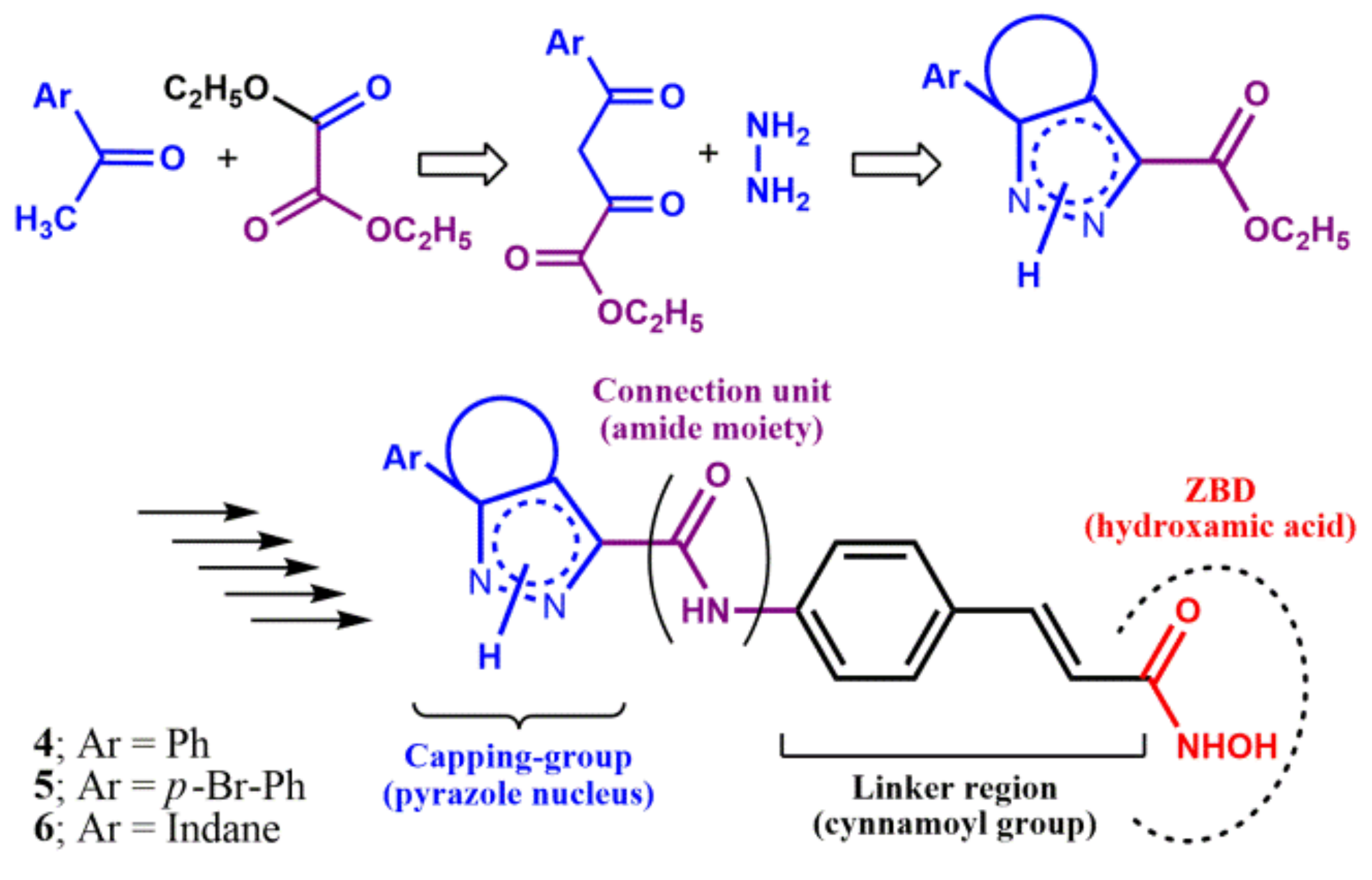
2.1. Chemistry
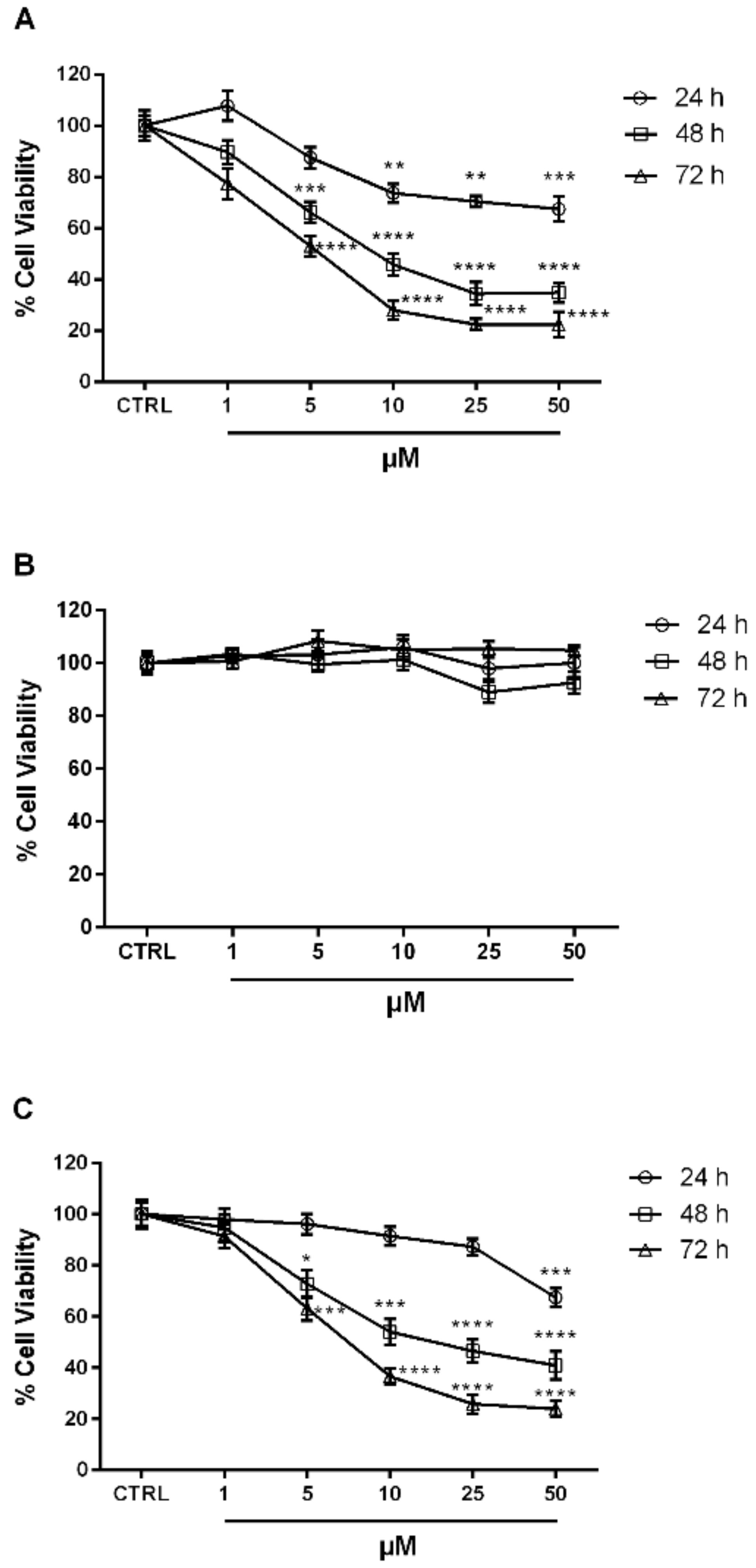
2.2. Biological Evaluation
2.3. Molecular Docking
3. Materials and Methods
3.1. Chemistry
3.1.1. General Experimental Information
3.1.2. General Procedure for the Synthesis of the N1-phenyl-pyrazole Scaffold
3.1.3. General Procedure for the Hydrolysis of the N1-phenyl-pyrazole Scaffold
3.1.4. General Procedure for the Coupling between the N1-phenyl-pyrazole Scaffold (Cap) and the Cinnamoyl Linker
3.1.5. General Procedure for the Hydrolysis of the Adduct N1-phenyl-pyrazole (Cap)/Cinnamoyl Linker
3.1.6. General Procedure for the Synthesis of the Final Cinnamoyl Hydroxamic Acid with CAP N1-phenyl-pyrazole (1a,1b)
3.1.7. Synthesis of the Other Hydroxamic Acids with CAP N1-aryl-pyrazole (2,3)
3.1.8. Synthesis of the α,γ-Diketoester Intermediate 26 and 27
3.1.9. Synthesis of N1H-pyrazole Scaffold C5-phenyl-substituted
3.1.10. Hydrolysis of the N1H-pyrazole Scaffolds C5-phenyl-substituted
3.1.11. Coupling Reaction between N1H-5-phenyl-pyrazole Scaffold (CAP) and Cinnamoyl Linker
3.1.12. Hydrolysis of the Adduct N1H-pyrazole CAP/Cinnamoyl Linker
3.1.13. Synthesis of the Final Hydroxamic Acid with CAP N1H-5-phenyl-pyrazole (4)
3.1.14. Synthesis of the Other Hydroxamic Acids with CAP N1H-aryl-substituted-pyrazole (5, 6)
3.2. Biological Activity
3.2.1. HDAC Inhibitor Drug Screening Kit
3.2.2. Cell Cultures and Treatment
3.2.3. Antiproliferative Activity
3.2.4. Statistical Analysis
3.3. Docking Studies
Docking Protocol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| EDCI | N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride |
| HATs | histone acetyltransferases |
| HDACIs | histone deacetylase inhibitors |
| HDACs | histone deacetylases |
| HOBt | hydroxybenzotriazole |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PDB | protein data bank |
| SAHA | suberoylanilide hydroxamic acid |
| TBDMSiO-NH2 | O-(tert-butyldimethylsilyl)hydroxylamine |
| TSA | Trichostatin A |
| ZBD | zinc-binding domain |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.M.; Zhu, W.G. Targeting histone deacetylases for cancer therapy: From molecular mechanisms to clinical implications. Int. J. Biol. Sci. 2014, 10, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Zagni, C.; Floresta, G.; Monciino, G.; Rescifina, A. The search for potent, small-molecule HDACIs in cancer treatment: A decade after vorinostat. Med. Res. Rev. 2017, 37, 1373–1428. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Rajak, H.; Singh, A.; Raghuwanshi, K.; Kumar, R.; Dewangan, P.K.; Veerasamy, R.; Sharma, P.C.; Dixit, A.; Mishra, P. A structural insight into hydroxamic acid based histone deacetylase inhibitors for the presence of anticancer activity. Curr. Med. Chem. 2014, 21, 2642–2664. [Google Scholar] [CrossRef]
- Bouchain, G.; Leit, S.; Frechette, S.; Abou Khalil, E.; Lavoie, R.; Moradei, O.; Woo, S.H.; Fournel, M.; Yan, P.T.; Kalita, A.; et al. Development of potential antitumor agents. Synthesis and biological evaluation of a new set of sulfonamide derivatives as histone deacetylase inhibitors. J. Med. Chem. 2003, 46, 820–830. [Google Scholar] [CrossRef]
- Mai, A.; Massa, S.; Pezzi, R.; Simeoni, S.; Rotili, D.; Nebbioso, A.; Scognamiglio, A.; Altucci, L.; Loidl, P.; Brosch, G. Class II (IIa)-selective histone deacetylase inhibitors. 1. Synthesis and biological evaluation of novel (aryloxopropenyl) pyrrolyl hydroxyamides. J. Med. Chem. 2005, 48, 3344–3353. [Google Scholar] [CrossRef]
- Chen, L.Q.; Petrelli, R.; Gao, G.Y.; Wilson, D.J.; McLean, G.T.; Jayaram, H.N.; Sham, Y.Y.; Pankiewicz, K.W. Dual inhibitors of inosine monophosphate dehydrogenase and histone deacetylase based on a cinnamic hydroxamic acid core structure. Bioorg. Med. Chem. 2010, 18, 5950–5964. [Google Scholar] [CrossRef]
- Valente, S.; Tardugno, M.; Conte, M.; Cirilli, R.; Perrone, A.; Ragno, R.; Simeoni, S.; Tramontano, A.; Massa, S.; Nebbioso, A.; et al. Novel cinnamyl hydroxyamides and 2-aminoanilides as histone deacetylase inhibitors: Apoptotic induction and cytodifferentiation activity. ChemMedChem 2011, 6, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Li, S.S.; Li, X.G.; Zhang, J.; Xu, W.F.; Li, X.C. Design, synthesis and preliminary biological evaluation of indoline-2,3-dione derivatives as novel HDAC inhibitors. Bioorg. Med. Chem. 2015, 23, 4728–4736. [Google Scholar] [CrossRef] [PubMed]
- He, S.P.; Dong, G.Q.; Wu, S.C.; Fang, K.; Miao, Z.Y.; Wang, W.; Sheng, C.Q. Small molecules simultaneously inhibiting p53-murine double minute 2 (MDM2) interaction and histone deacetylases (HDACs): Discovery of novel multitargeting antitumor agents. J. Med. Chem. 2018, 61, 7245–7260. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.C.; Niu, Q.; Liu, J.; Bao, Y.; Yang, J.Y.; Luan, S.L.; Fan, Y.B.; Liu, D.; Zhao, L.X. Novel thiol-based histone deacetylase inhibitors bearing 3-phenyl-1H-pyrazole-5-carboxamide scaffold as surface recognition motif: Design, synthesis and SAR study. Bioorg. Med. Chem. Lett. 2016, 26, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Cheng, G.L.; Xu, Q.H.; Luan, S.L.; Wang, S.X.; Liu, D.; Zhao, L.X. Design, synthesis and biological evaluation of novel hydroxamic acid based histone deacetylase 6 selective inhibitors bearing phenylpyrazol scaffold as surface recognition motif. Bioorg. Med. Chem. 2018, 26, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Pommery, N.; Taverne, T.; Telliez, A.; Goossens, L.; Charlier, C.; Pommery, J.; Goossens, J.F.; Houssin, R.; Durant, F.; Henichart, J.P. New COX-2/5-LOX inhibitors: Apoptosis-inducing agents potentially useful in prostate cancer chemotherapy. J. Med. Chem. 2004, 47, 6195–6206. [Google Scholar] [CrossRef] [PubMed]
- Nuti, E.; Santamaria, S.; Casalini, F.; Yamamoto, K.; Marinelli, L.; La Pietra, V.; Novellino, E.; Orlandini, E.; Nencetti, S.; Marini, A.M.; et al. Arylsulfonamide inhibitors of aggrecanases as potential therapeutic agents for osteoarthritis: Synthesis and biological evaluation. Eur. J. Med. Chem. 2013, 62, 379–394. [Google Scholar] [CrossRef]
- Van Herk, T.; Brussee, J.; van den Nieuwendijk, A.M.C.H.; van der Klein, P.A.M.; IJzerman, A.P.; Stannek, C.; Burmeister, A.; Lorenzen, A. Pyrazole derivatives as partial Agonists for the nicotinic acid receptor. J. Med. Chem. 2003, 46, 3945–3951. [Google Scholar] [CrossRef]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [Google Scholar] [CrossRef]
- Navarra, M.; Ferlazzo, N.; Cirmi, S.; Trapasso, E.; Bramanti, P.; Lombardo, G.E.; Minciullo, P.L.; Calapai, G.; Gangemi, S. Effects of bergamot essential oil and its extractive fractions on SH-SY5Y human neuroblastoma cell growth. J. Pharm. Pharmacol. 2015, 67, 1042–1053. [Google Scholar] [CrossRef]
- Ferlazzo, N.; Cirmi, S.; Russo, M.; Trapasso, E.; Ursino, M.R.; Lombardo, G.E.; Gangemi, S.; Calapai, G.; Navarra, M. NF-kappa B mediates the antiproliferative and proapoptotic effects of bergamot juice in HepG2 cells. Life Sci. 2016, 146, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, N.; Seto, E. Regulation of histone deacetylase activities. J. Cell. Biochem. 2004, 93, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA—An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Aided Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4(Zn): An improved AutoDock force field for small-molecule docking to zinc metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
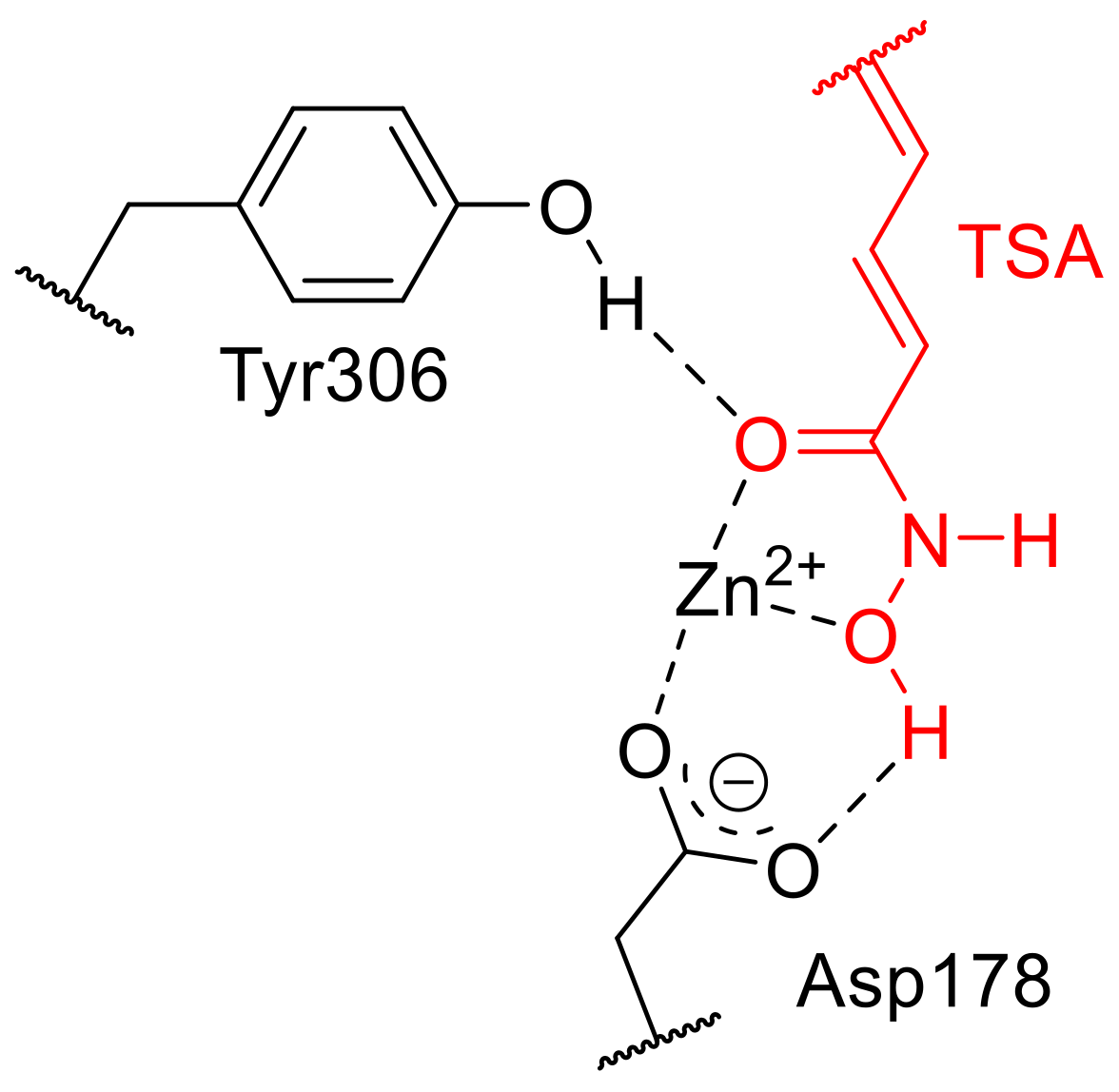{kind=link}
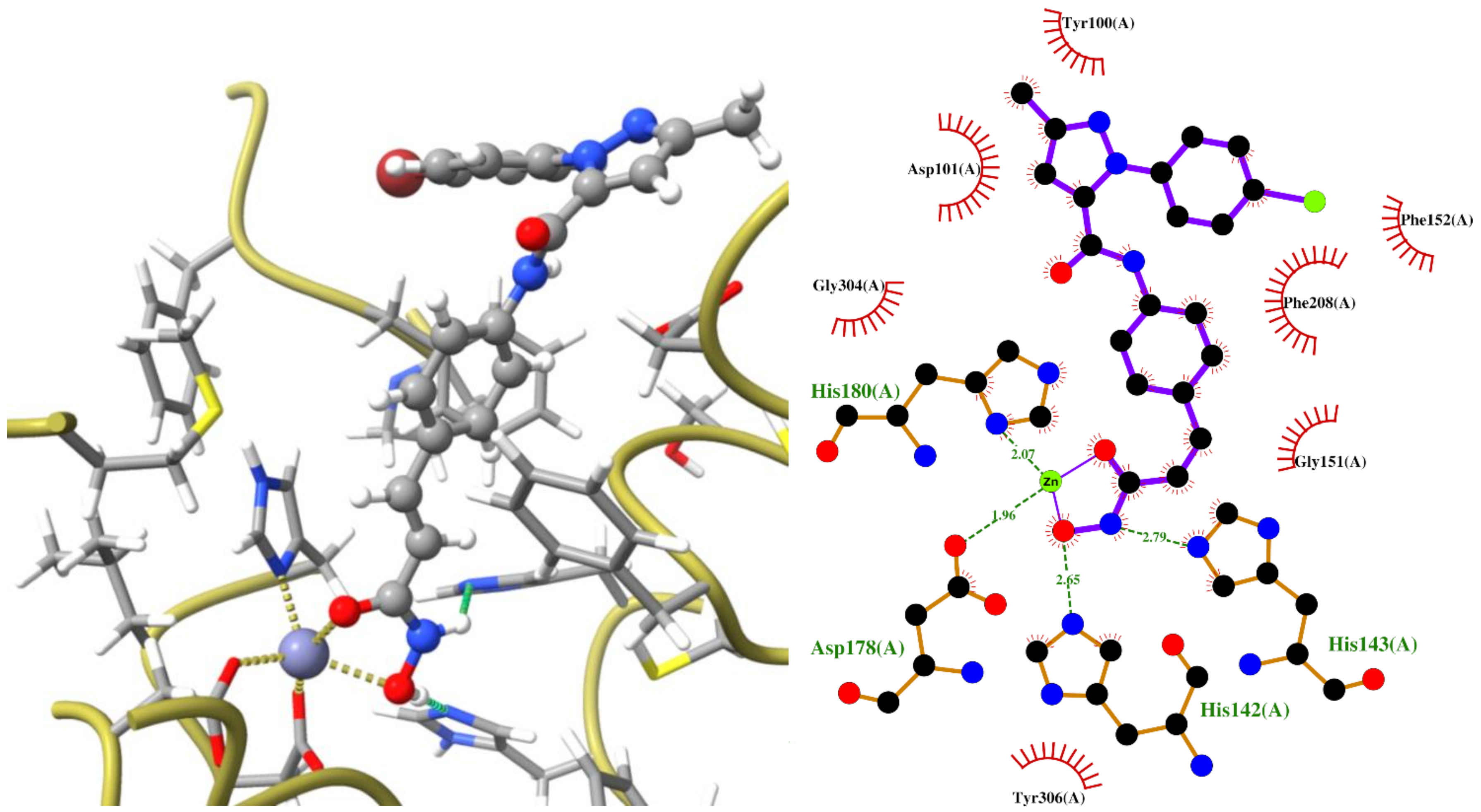{kind=link}
| Compd. | Inhibition at 10 μM (%) a | Inhibition at 500 μM (%) a | IC50 ±SD b (μM) | Calcd ΔGbind c | Calcd Ki (μM) |
|---|---|---|---|---|---|
| 1a | — | 86 | 7.1 ± 0.5 | −7.96 | 1.45 |
| 1b | 6 | 30 | >500 | −6.04 | 37.19 |
| 2 | 51 | 88 | 6.3 ± 0.3 | −8.08 | 1.19 |
| 3 | 74 | 88 | 1.6 ± 0.2 | −8.79 | 0.36 |
| 4 | 13 | 71 | 155.2 ± 2.8 | −6.40 | 20.25 |
| 5 | — | 15 | >500 | −5.53 | 87.99 |
| 6 | 29 | 87 | 46.5 ± 1.5 | −7.27 | 4.69 |
| TSA d | — | — | 3.4 ± 0.2 | −11.13 | 0.007 |
| SAHA d | — | — | — | −10.03 | 0.044 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zagni, C.; Citarella, A.; Oussama, M.; Rescifina, A.; Maugeri, A.; Navarra, M.; Scala, A.; Piperno, A.; Micale, N. Hydroxamic Acid-Based Histone Deacetylase (HDAC) Inhibitors Bearing a Pyrazole Scaffold and a Cinnamoyl Linker. Int. J. Mol. Sci. 2019, 20, 945. https://doi.org/10.3390/ijms20040945
Zagni C, Citarella A, Oussama M, Rescifina A, Maugeri A, Navarra M, Scala A, Piperno A, Micale N. Hydroxamic Acid-Based Histone Deacetylase (HDAC) Inhibitors Bearing a Pyrazole Scaffold and a Cinnamoyl Linker. International Journal of Molecular Sciences. 2019; 20(4):945. https://doi.org/10.3390/ijms20040945
Chicago/Turabian StyleZagni, Chiara, Andrea Citarella, Mahjoub Oussama, Antonio Rescifina, Alessandro Maugeri, Michele Navarra, Angela Scala, Anna Piperno, and Nicola Micale. 2019. "Hydroxamic Acid-Based Histone Deacetylase (HDAC) Inhibitors Bearing a Pyrazole Scaffold and a Cinnamoyl Linker" International Journal of Molecular Sciences 20, no. 4: 945. https://doi.org/10.3390/ijms20040945
APA StyleZagni, C., Citarella, A., Oussama, M., Rescifina, A., Maugeri, A., Navarra, M., Scala, A., Piperno, A., & Micale, N. (2019). Hydroxamic Acid-Based Histone Deacetylase (HDAC) Inhibitors Bearing a Pyrazole Scaffold and a Cinnamoyl Linker. International Journal of Molecular Sciences, 20(4), 945. https://doi.org/10.3390/ijms20040945