Modulation of Obesity and Insulin Resistance by the Redox Enzyme and Adaptor Protein p66Shc



Abstract
:1. Introduction
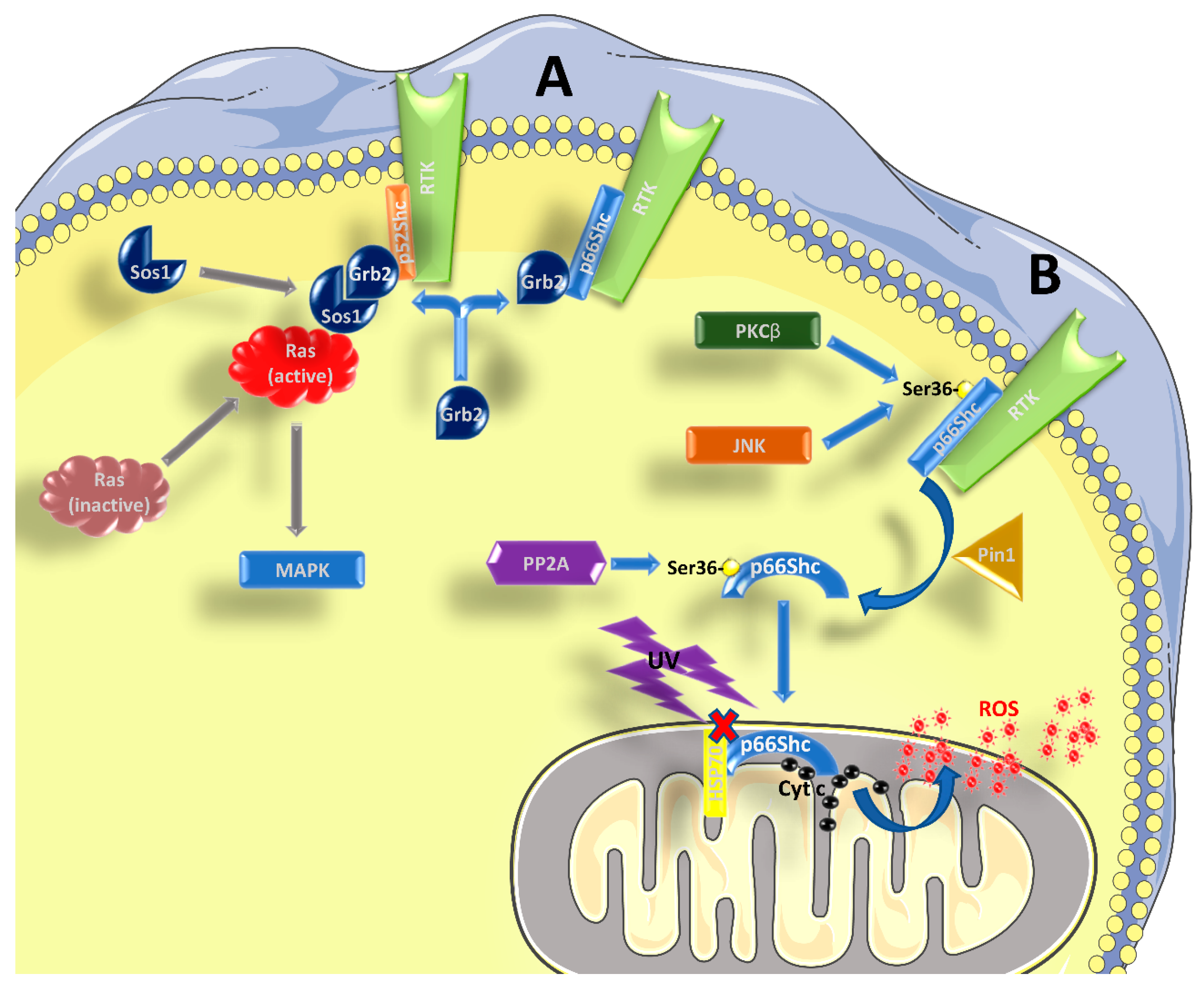
1.1. Role of p66Shc in Signal Transduction
1.2. p66Shc and Longevity
1.3. p66Shc, Body Weight Regulation, and Obesity
1.4. p66Shc, Diabetes, and the IGF-1 Axis
1.5. Role of p66Shc in the Regulation of Glucose Homeostasis
1.6. Body Temperature Regulation, Respiration, and Energy Expenditure
2. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Pelicci, G.; Lanfrancone, L.; Grignani, F.; McGlade, J.; Cavallo, F.; Forni, G.; Nicoletti, I.; Grignani, F.; Pawson, T.; Pelicci, P.G. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 1992, 70, 93–104. [Google Scholar] [CrossRef]
- Migliaccio, E.; Mele, S.; Salcini, A.E.; Pelicci, G.; Lai, K.M.; Superti-Furga, G.; Pawson, T.; Di Fiore, P.P.; Lanfrancone, L.; Pelicci, P.G. Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J. 1997, 16, 706–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luzi, L.; Confalonieri, S.; Di Fiore, P.P.; Pelicci, P.G. Evolution of Shc functions from nematode to human. Curr. Opin. Genet. Dev. 2000, 10, 668–674. [Google Scholar] [CrossRef]
- Bhat, S.S.; Anand, D.; Khanday, F.A. p66Shc as a switch in bringing about contrasting responses in cell growth: Implications on cell proliferation and apoptosis. Mol. Cancer 2015, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Galimov, E.R. The Role of p66shc in Oxidative Stress and Apoptosis. Acta Nat. 2010, 2, 44–51. [Google Scholar]
- Bonfini, L.; Migliaccio, E.; Pelicci, G.; Lanfrancone, L.; Pelicci, P.G. Not all Shc’s roads lead to Ras. Trends Biochem. Sci. 1996, 21, 257–261. [Google Scholar] [CrossRef]
- Okada, S.; Kao, A.W.; Ceresa, B.P.; Blaikie, P.; Margolis, B.; Pessin, J.E. The 66-kDa Shc isoform is a negative regulator of the epidermal growth factor-stimulated mitogen-activated protein kinase pathway. J. Biol. Chem. 1997, 272, 28042–28049. [Google Scholar] [CrossRef] [PubMed]
- Pacini, S.; Pellegrini, M.; Migliaccio, E.; Patrussi, L.; Ulivieri, C.; Ventura, A.; Carraro, F.; Naldini, A.; Lanfrancone, L.; Pelicci, P.; et al. p66SHC promotes apoptosis and antagonizes mitogenic signaling in T cells. Mol. Cell. Biol. 2004, 24, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Shen, X.; Clemmons, D.R. p66shc negatively regulates insulin-like growth factor I signal transduction via inhibition of p52shc binding to Src homology 2 domain-containing protein tyrosine phosphatase substrate-1 leading to impaired growth factor receptor-bound protein-2 membrane recruitment. Mol. Endocrinol. 2008, 22, 2162–2175. [Google Scholar] [PubMed]
- Arany, I.; Faisal, A.; Nagamine, Y.; Safirstein, R.L. p66shc inhibits pro-survival epidermal growth factor receptor/ERK signaling during severe oxidative stress in mouse renal proximal tubule cells. J. Biol. Chem. 2008, 283, 6110–6117. [Google Scholar] [CrossRef] [PubMed]
- Khanday, F.A.; Santhanam, L.; Kasuno, K.; Yamamori, T.; Naqvi, A.; Dericco, J.; Bugayenko, A.; Mattagajasingh, I.; Disanza, A.; Scita, G.; et al. Sos-mediated activation of rac1 by p66shc. J. Cell Biol. 2006, 172, 817–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliaccio, E.; Giorgio, M.; Mele, S.; Pelicci, G.; Reboldi, P.; Pandolfi, P.P.; Lanfrancone, L.; Pelicci, P.G. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999, 402, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Rizzuto, R. p66Shc, oxidative stress and aging: Importing a lifespan determinant into mitochondria. Cell Cycle 2008, 7, 304–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natalicchio, A.; Tortosa, F.; Perrini, S.; Laviola, L.; Giorgino, F. p66Shc, a multifaceted protein linking Erk signalling, glucose metabolism, and oxidative stress. Arch. Physiol. Biochem. 2011, 117, 116–124. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, E.; Baldassari, F.; Bononi, A.; Wieckowski, M.R.; Pinton, P. Oxidative stress in cardiovascular diseases and obesity: Role of p66Shc and protein kinase C. Oxid. Med. Cell. Longev. 2013, 2013, 564961. [Google Scholar] [CrossRef] [PubMed]
- Trinei, M.; Giorgio, M.; Cicalese, A.; Barozzi, S.; Ventura, A.; Migliaccio, E.; Milia, E.; Padura, I.M.; Raker, V.A.; Maccarana, M.; et al. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene 2002, 21, 3872–3878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinei, M.; Migliaccio, E.; Bernardi, P.; Paolucci, F.; Pelicci, P.; Giorgio, M. p66Shc, mitochondria, and the generation of reactive oxygen species. Methods Enzymol. 2013, 528, 99–110. [Google Scholar] [PubMed]
- Di Lisa, F.; Giorgio, M.; Ferdinandy, P.; Schulz, R. New aspects of p66Shc in ischemia reperfusion injury and cardiovascular diseases. Br. J. Pharmacol. 2017, 174, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Le, S.; Connors, T.J.; Maroney, A.C. c-Jun N-terminal kinase specifically phosphorylates p66ShcA at serine 36 in response to ultraviolet irradiation. J. Biol. Chem. 2001, 276, 48332–48336. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Rimessi, A.; Marchi, S.; Orsini, F.; Migliaccio, E.; Giorgio, M.; Contursi, C.; Minucci, S.; Mantovani, F.; Wieckowski, M.R.; et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 2007, 315, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Haller, M.; Khalid, S.; Kremser, L.; Fresser, F.; Furlan, T.; Hermann, M.; Guenther, J.; Drasche, A.; Leitges, M.; Giorgio, M.; et al. Novel Insights into the PKCbeta-dependent Regulation of the Oxidoreductase p66Shc. J. Biol. Chem. 2016, 291, 23557–23568. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Migliaccio, E.; Moroni, M.; Contursi, C.; Raker, V.A.; Piccini, D.; Martin-Padura, I.; Pelliccia, G.; Trinei, M.; Bono, M.; et al. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J. Biol. Chem. 2004, 279, 25689–25695. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Moroni, M.; Contursi, C.; Yano, M.; Pelicci, P.; Giorgio, M.; Migliaccio, E. Regulatory effects of the mitochondrial energetic status on mitochondrial p66Shc. Biol. Chem. 2006, 387, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Francia, P.; Camici, G.G.; Pelicci, P.G.; Luscher, T.F.; Volpe, M. Final common molecular pathways of aging and cardiovascular disease: Role of the p66Shc protein. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Starkov, A.A. Mitochondrial ROS Metabolism: 10 Years Later. Biochemistry (Mosc) 2015, 80, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Steegborn, C. The mitochondrial apoptosis pathway and p66Shc--a regulatory redox enzyme or an adapter protein snuggling around? Cell Cycle 2010, 9, 4425–4426. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, M.; Tsuji, Y. Evidence for a novel antioxidant function and isoform-specific regulation of the human p66Shc gene. Mol. Biol. Cell 2014, 25, 2116–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansone, P.; Storci, G.; Giovannini, C.; Pandolfi, S.; Pianetti, S.; Taffurelli, M.; Santini, D.; Ceccarelli, C.; Chieco, P.; Bonafe, M. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells 2007, 25, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Jung, S.B.; Naqvi, A.; Hoffman, T.A.; DeRicco, J.; Yamamori, T.; Cole, M.P.; Jeon, B.H.; Irani, K. p53 impairs endothelium-dependent vasomotor function through transcriptional upregulation of p66shc. Circ. Res. 2008, 103, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Marchi, S.; Rimessi, A.; Bonora, M.; Giorgi, C.; Mehta, K.D.; Pinton, P. PRKCB/protein kinase C, beta and the mitochondrial axis as key regulators of autophagy. Autophagy 2013, 9, 1367–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granatiero, V.; Gherardi, G.; Vianello, M.; Salerno, E.; Zecchini, E.; Toniolo, L.; Pallafacchina, G.; Murgia, M.; Blaauw, B.; Rizzuto, R.; et al. Role of p66shc in skeletal muscle function. Sci. Rep. 2017, 7, 6283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weindruch, R.; Walford, R.L. Dietary restriction in mice beginning at 1 year of age: Effect on life-span and spontaneous cancer incidence. Science 1982, 215, 1415–1418. [Google Scholar] [CrossRef] [PubMed]
- Weindruch, R.; Walford, R.L.; Fligiel, S.; Guthrie, D. The retardation of aging in mice by dietary restriction: Longevity, cancer, immunity and lifetime energy intake. J. Nutr. 1986, 116, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, J.J.; Tran, D.; Giorgio, M.; Griffey, S.M.; Koehne, A.; Laing, S.T.; Taylor, S.L.; Kim, K.; Cortopassi, G.A.; Lloyd, K.C.; et al. The influence of Shc proteins on life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Bonafe, M.; Di Tella, L.; Tiberi, L.; Salvioli, S.; Monti, D.; Sorbi, S.; Franceschi, C. p66(shc) is highly expressed in fibroblasts from centenarians. Mech. Ageing Dev. 2005, 126, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Stendardo, M.; Migliaccio, E.; Pelicci, P.G. P66SHC deletion improves fertility and progeric phenotype of late-generation TERC-deficient mice but not their short lifespan. Aging Cell 2016, 15, 446–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berniakovich, I.; Trinei, M.; Stendardo, M.; Migliaccio, E.; Minucci, S.; Bernardi, P.; Pelicci, P.G.; Giorgio, M. p66Shc-generated oxidative signal promotes fat accumulation. J Biol Chem 2008, 283, 34283–34293. [Google Scholar] [CrossRef] [PubMed]
- Ciciliot, S.; Albiero, M.; Menegazzo, L.; Poncina, N.; Scattolini, V.; Danesi, A.; Pagnin, E.; Marabita, M.; Blaauw, B.; Giorgio, M.; et al. p66Shc deletion or deficiency protects from obesity but not metabolic dysfunction in mice and humans. Diabetologia 2015, 58, 2352–2360. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.H.; Kim, K.; Ramsey, J.J. The influence of shc proteins and aging on whole body energy expenditure and substrate utilization in mice. PLoS ONE 2012, 7, e48790. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.H.; Kim, K.; Ramsey, J.J. The influence of acute, late-life calorie restriction on whole body energy metabolism in p66Shc(−/−) mice. Mech. Ageing Dev. 2012, 133, 414–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranieri, S.C.; Fusco, S.; Panieri, E.; Labate, V.; Mele, M.; Tesori, V.; Ferrara, A.M.; Maulucci, G.; De Spirito, M.; Martorana, G.E.; et al. Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 13420–13425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciciliot, S.; Albiero, M.; Campanaro, S.; Poncina, N.; Tedesco, S.; Scattolini, V.; Dalla Costa, F.; Cignarella, A.; Vettore, M.; Di Gangi, I.M.; et al. Interplay between gut microbiota and p66Shc affects obesity-associated insulin resistance. FASEB J. 2018, 32, 4004–4015. [Google Scholar] [CrossRef] [PubMed]
- Tomilov, A.A.; Ramsey, J.J.; Hagopian, K.; Giorgio, M.; Kim, K.M.; Lam, A.; Migliaccio, E.; Lloyd, K.C.; Berniakovich, I.; Prolla, T.A.; et al. The Shc locus regulates insulin signaling and adiposity in mammals. Aging Cell 2011, 10, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Berry, A.; Berniakovich, I.; Poletaeva, I.; Trinei, M.; Stendardo, M.; Hagopian, K.; Ramsey, J.J.; Cortopassi, G.; Migliaccio, E.; et al. The p66Shc knocked out mice are short lived under natural condition. Aging Cell 2012, 11, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Bellisario, V.; Berry, A.; Capoccia, S.; Raggi, C.; Panetta, P.; Branchi, I.; Piccaro, G.; Giorgio, M.; Pelicci, P.G.; Cirulli, F. Gender-dependent resiliency to stressful and metabolic challenges following prenatal exposure to high-fat diet in the p66(Shc−/−) mouse. Front. Behav. Neurosci. 2014, 8, 285. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Ceolotto, G.; Pagnin, E.; de Kreutzenberg, S.; Avogaro, A. At the crossroads of longevity and metabolism: The metabolic syndrome and lifespan determinant pathways. Aging Cell 2011, 10, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Chen, H.Z.; Wan, Y.Z.; Zhang, Q.J.; Wei, Y.S.; Huang, S.; Liu, J.J.; Lu, Y.B.; Zhang, Z.Q.; Yang, R.F.; et al. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ. Res. 2011, 109, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kim, Y.R.; Vikram, A.; Kumar, S.; Kassan, M.; Gabani, M.; Lee, S.K.; Jacobs, J.S.; Irani, K. P66Shc-Induced MicroRNA-34a Causes Diabetic Endothelial Dysfunction by Downregulating Sirtuin1. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2394–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rota, M.; LeCapitaine, N.; Hosoda, T.; Boni, A.; De Angelis, A.; Padin-Iruegas, M.E.; Esposito, G.; Vitale, S.; Urbanek, K.; Casarsa, C.; et al. Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circ. Res. 2006, 99, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; Pagnin, E.; Iori, E.; Cosma, C.; Lapolla, A.; Pengo, V.; Stendardo, M.; et al. The redox enzyme p66Shc contributes to diabetes and ischemia-induced delay in cutaneous wound healing. Diabetes 2010, 59, 2306–2314. [Google Scholar] [CrossRef] [PubMed]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Natalicchio, A.; Laviola, L.; De Tullio, C.; Renna, L.A.; Montrone, C.; Perrini, S.; Valenti, G.; Procino, G.; Svelto, M.; Giorgino, F. Role of the p66Shc isoform in insulin-like growth factor I receptor signaling through MEK/Erk and regulation of actin cytoskeleton in rat myoblasts. J. Biol. Chem. 2004, 279, 43900–43909. [Google Scholar] [CrossRef] [PubMed]
- Natalicchio, A.; De Stefano, F.; Perrini, S.; Laviola, L.; Cignarelli, A.; Caccioppoli, C.; Quagliara, A.; Melchiorre, M.; Leonardini, A.; Conserva, A.; et al. Involvement of the p66Shc protein in glucose transport regulation in skeletal muscle myoblasts. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E228–E237. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z. Adapter proteins regulate insulin resistance and lipid metabolism in obesity. Sci. Bull. 2016, 61, 8. [Google Scholar] [CrossRef]
- Ranieri, S.C.; Fusco, S.; Pani, G. p66(ShcA): Linking mammalian longevity with obesity-induced insulin resistance. Vitam. Horm. 2013, 91, 219–241. [Google Scholar] [PubMed]
- Soliman, M.A.; Abdel Rahman, A.M.; Lamming, D.W.; Birsoy, K.; Pawling, J.; Frigolet, M.E.; Lu, H.; Fantus, I.G.; Pasculescu, A.; Zheng, Y.; et al. The adaptor protein p66Shc inhibits mTOR-dependent anabolic metabolism. Sci. Signal. 2014, 7, ra17. [Google Scholar] [CrossRef] [PubMed]
- Hagopian, K.; Tomilov, A.A.; Kim, K.; Cortopassi, G.A.; Ramsey, J.J. Key glycolytic enzyme activities of skeletal muscle are decreased under fed and fasted states in mice with knocked down levels of Shc proteins. PLoS ONE 2015, 10, e0124204. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Combs, C.A.; French, S.; Ahn, B.H.; Fergusson, M.M.; Balaban, R.S.; Finkel, T. The mammalian longevity-associated gene product p66shc regulates mitochondrial metabolism. J. Biol. Chem. 2006, 281, 10555–10560. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.N.; Barker, D.J. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Condition | In Favor | Against |
|---|---|---|
| Increased longevity | Deletion of p66Shc increases average and maximum longevity [12] | No increase in lifespan of p66Shc−/− mice [35] High expression of p66Shc in centenarians [36] Deletion of p66Shc does not rescue TREC deficiency [37] |
| Bodyweight regulation | p66Shc−/− mice are leaner than age-matched WT [35,38,39,40,43] | p66Shc deletion has no influence on bodyweight [12,40,41] |
| Protection from obesity | Knockout mice gain less weight than WT controls [38,39,42,43] | Knockout mice gain more weight than WT controls [44] |
| Improved glucose homeostasis | Glucose uptake is increased in the absence of p66Shc [42,44,54,57,58] Increased glycolysis [57,59] Improved glucose tolerance and/or insulin sensitivity in lean [44] and obese knockout animals [42] | Glucose uptake is not increased [42] or even decreased [39,44] in the absence of p66Shc Decreased glycolysis [58] Similar or worsened glucose tolerance and/or insulin sensitivity in lean [39,43,44] and obese knockout animals [39,43,44] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciciliot, S.; Fadini, G.P. Modulation of Obesity and Insulin Resistance by the Redox Enzyme and Adaptor Protein p66Shc. Int. J. Mol. Sci. 2019, 20, 985. https://doi.org/10.3390/ijms20040985
Ciciliot S, Fadini GP. Modulation of Obesity and Insulin Resistance by the Redox Enzyme and Adaptor Protein p66Shc. International Journal of Molecular Sciences. 2019; 20(4):985. https://doi.org/10.3390/ijms20040985
Chicago/Turabian StyleCiciliot, Stefano, and Gian Paolo Fadini. 2019. "Modulation of Obesity and Insulin Resistance by the Redox Enzyme and Adaptor Protein p66Shc" International Journal of Molecular Sciences 20, no. 4: 985. https://doi.org/10.3390/ijms20040985
APA StyleCiciliot, S., & Fadini, G. P. (2019). Modulation of Obesity and Insulin Resistance by the Redox Enzyme and Adaptor Protein p66Shc. International Journal of Molecular Sciences, 20(4), 985. https://doi.org/10.3390/ijms20040985