Molecular Mechanisms of Hypothalamic Insulin Resistance

{kind=link}
Abstract
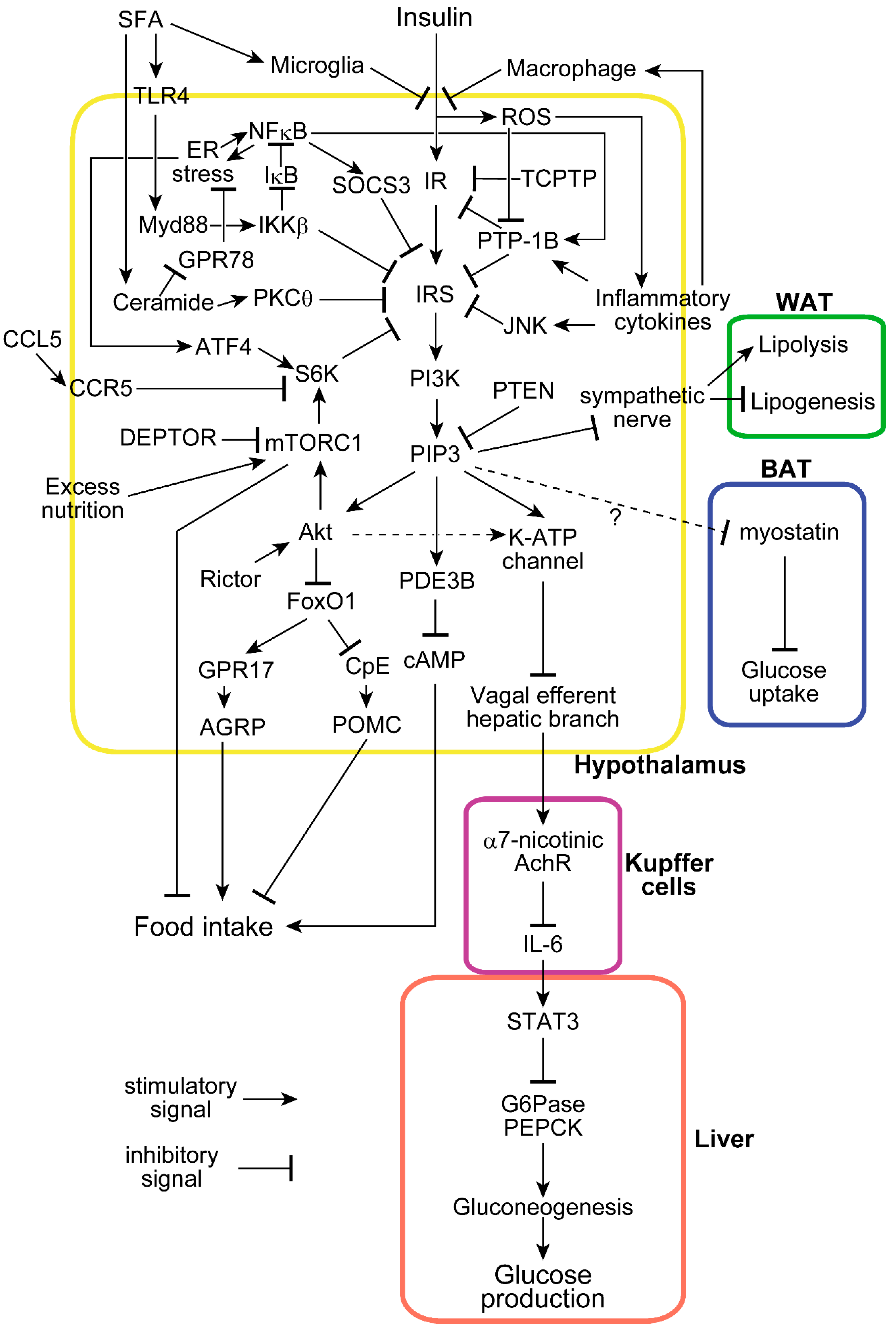
:1. Introduction
2. Two Major Insulin Functions in the Hypothalamus: Suppression of Food Intake and Endogenous Glucose Production
3. Hypothalamic Insulin Resistance Induced by Excessive Nutrition
4. Inflammation with ER Stress Induces Hypothalamic Insulin Resistance
5. Involvement of Phosphatases and SOCS3 in Hypothalamic Insulin Resistance
6. Involvement of the mTOR-S6 Kinase Pathway in Hypothalamic Insulin Resistance
7. Cell Populations Involved in Hypothalamic Insulin Functions and Insulin Resistance
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AchR | Acetylcholine receptor |
| AGRP | Agouti-related protein |
| ATF4 | Activating transcription factor 4 |
| BAT | Brown adipose tissue |
| BBB | Blood–brain barrier |
| CCL5 | C C motif chemokine 5 |
| CCR5 | C–C chemokine receptor type 5 |
| CpE | Carboxypeptidase E |
| CSF | Cerebrospinal fluid |
| DEPTOR | DEP domain-containing mTOR-interacting protein |
| DREADD | Designer receptors exclusively activated by designer drugs |
| EGP | Endogenous glucose production |
| ER stress | Endoplasmic reticulum stress |
| FoxO1 | Forkhead box protein O1 |
| G6Pase | Glucose 6-phosphatase |
| GPR | G protein-coupled receptor |
| HFD | High-fat diet |
| IκB | Nuclear factor of kappa-light-chain-enhancer in B-cells inhibitor |
| IKKβ | IκB kinase β |
| IL-6 | Interleukin 6 |
| IR | Insulin receptor |
| IRS | Insulin receptor substrate |
| JNK | c-Jun-NH2-terminal kinase |
| mTORC1 | Mechanistic target of rapamycin complex 1 |
| Myd88 | Myeloid differentiation primary response 88 |
| NFκB | Nuclear factor of kappa-light-chain-enhancer in B-cells |
| NPY | Neuropeptide Y |
| PDE3B | Phosphodiesterase 3B |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PI3K | Phosphoinositide 3-kinase |
| PIP3 | Phosphatidylinositol 3,4,5-triphosphate |
| PKC | Protein kinase C |
| POMC | Proopiomelanocortin |
| PTEN | Phosphatase and tensin homolog on chromosome 10 |
| PTP-1B | Protein tyrosine phosphatase 1B |
| RANTES | Regulated on activation, normal T cell expressed and secreted |
| Rictor | Rapamycin-insensitive companion of mammalian target of rapamycin |
| ROS | Reactive oxygen species |
| S6K | P70 S6-kinase |
| SFA | Saturated fatty acids |
| STAT3 | Signal transducer and activator of transcription 3 |
| TCPTP | T-cell protein tyrosine phosphatase |
| WAT | White adipose tissue |
References
- Yanovski, S.Z.; Yanovski, J.A. Long-term Drug Treatment for Obesity: A Systematic and Clinical Review. JAMA 2014, 311, 74–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, S.C.; Lotter, E.C.; McKay, L.D.; Porte, D. Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 1979, 282, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Carey, M.; Kehlenbrink, S.; Hawkins, M. Evidence for Central Regulation of Glucose Metabolism. J. Biol. Chem. 2013, 288, 34981–34988. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Tanida, M.; Nagata, N.; Inaba, Y.; Watanabe, H.; Nagashimada, M.; Ota, T.; Asahara, S.; Kido, Y.; Matsumoto, M.; et al. Central Insulin Action Activates Kupffer Cells by Suppressing Hepatic Vagal Activation via the Nicotinic Alpha 7 Acetylcholine Receptor. Cell Rep. 2016, 14, 2362–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedict, C.; Kern, W.; Schultes, B.; Born, J.; Hallschmid, M. Differential Sensitivity of Men and Women to Anorexigenic and Memory-Improving Effects of Intranasal Insulin. J. Clin. Endocrinol. Metab. 2008, 93, 1339–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heni, M.; Wagner, R.; Kullmann, S.; Gancheva, S.; Roden, M.; Peter, A.; Stefan, N.; Preissl, H.; Häring, H.-U.; Fritsche, A. Hypothalamic and Striatal Insulin Action Suppresses Endogenous Glucose Production and May Stimulate Glucose Uptake During Hyperinsulinemia in Lean but Not in Overweight Men. Diabetes 2017, 66, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Brüning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Müller-Wieland, D.; Kahn, C.R. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef] [PubMed]
- Obici, S.; Feng, Z.; Karkanias, G.; Baskin, D.G.; Rossetti, L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat. Neurosci. 2002, 5, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin Action in Brain Regulates Systemic Metabolism and Brain Function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, A.M.; Hernandez-Garzón, E.; Perez-Domper, P.; Perez-Alvarez, A.; Mederos, S.; Matsui, T.; Santi, A.; Trueba-Saiz, A.; García-Guerra, L.; Pose-Utrilla, J.; et al. Insulin Regulates Astrocytic Glucose Handling Through Cooperation With IGF-I. Diabetes 2017, 66, 64–74. [Google Scholar] [CrossRef]
- García-Cáceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.-X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.M.; Aylor, K.W.; Barrett, E.J. Unravelling the regulation of insulin transport across the brain endothelial cell. Diabetologia 2017, 60, 1512–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, R.I.; Gray, S.M.; Aylor, K.W.; Barrett, E.J. Pathways for insulin access to the brain: The role of the microvascular endothelial cell. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1132–H1138. [Google Scholar] [CrossRef] [PubMed]
- Konishi, M.; Sakaguchi, M.; Lockhart, S.M.; Cai, W.; Li, M.E.; Homan, E.P.; Rask-Madsen, C.; Kahn, C.R. Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc. Natl. Acad. Sci. USA 2017, 114, E8478–E8487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, E.M.; Blázquez, J.L.; Guerra, M. The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: The former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides 2010, 31, 757–776. [Google Scholar] [CrossRef] [PubMed]
- Balland, E.; Dam, J.; Langlet, F.; Caron, E.; Steculorum, S.; Messina, A.; Rasika, S.; Falluel-Morel, A.; Anouar, Y.; Dehouck, B.; et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab. 2014, 19, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Tschöp, M.H.; Luquet, S. Hypothalamic Tanycytes: Gatekeepers to Metabolic Control. Cell Metab. 2014, 19, 173–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, G.T.; Tiganis, T. Insulin action in the brain: Roles in energy and glucose homeostasis. J. Neuroendocrinol. 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Alegría, K.; Flores-León, M.; Avila-Muñoz, E.; Rodríguez-Corona, N.; Arias, C. PI3K Signaling in Neurons: A Central Node for the Control of Multiple Functions. Int. J. Mol. Sci. 2018, 19, 3725. [Google Scholar] [CrossRef]
- Niswender, K.D.; Morrison, C.D.; Clegg, D.J.; Olson, R.; Baskin, D.G.; Myers, M.G.; Seeley, R.J.; Schwartz, M.W. Insulin Activation of Phosphatidylinositol 3-Kinase in the Hypothalamic Arcuate Nucleus: A Key Mediator of Insulin-Induced Anorexia. Diabetes 2003, 52, 227–231. [Google Scholar] [CrossRef]
- Gelling, R.W.; Morton, G.J.; Morrison, C.D.; Niswender, K.D.; Myers, M.G.; Rhodes, C.J.; Schwartz, M.W. Insulin action in the brain contributes to glucose lowering during insulin treatment of diabetes. Cell Metab. 2006, 3, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumita, T.; Ono, H.; Suzuki, T.; Sakai, G.; Inukai, K.; Katagiri, H.; Asano, T.; Katayama, S.; Awata, T. Mediobasal hypothalamic PTEN modulates hepatic insulin resistance independently of food intake in rats. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E47–E60. [Google Scholar] [CrossRef] [PubMed]
- Kocalis, H.E.; Hagan, S.L.; George, L.; Turney, M.K.; Siuta, M.A.; Laryea, G.N.; Morris, L.C.; Muglia, L.J.; Printz, R.L.; Stanwood, G.D.; et al. Rictor/mTORC2 facilitates central regulation of energy and glucose homeostasis. Mol. Metab. 2014, 3, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Orozco, I.J.; Su, Y.; Suyama, S.; Gutiérrez-Juárez, R.; Horvath, T.L.; Wardlaw, S.L.; Plum, L.; Arancio, O.; Accili, D. FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell 2012, 149, 1314–1326. [Google Scholar] [CrossRef] [PubMed]
- Plum, L.; Lin, H.V.; Dutia, R.; Tanaka, J.; Aizawa, K.S.; Matsumoto, M.; Kim, A.J.; Cawley, N.X.; Paik, J.; Loh, Y.P.; et al. The Obesity Susceptibility Gene Carboxypeptidase E Links FoxO1 Signaling in Hypothalamic Pro–opiomelanocortin Neurons with Regulation of Food Intake. Nat. Med. 2009, 15, 1195–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, G.; Meece, K.; Wardlaw, S.L.; Accili, D. Preserved energy balance in mice lacking FoxO1 in neurons of Nkx2.1 lineage reveals functional heterogeneity of FoxO1 signaling within the hypothalamus. Diabetes 2014, 63, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Ogawa, W.; Asakawa, A.; Okamoto, Y.; Nishizawa, A.; Matsumoto, M.; Teshigawara, K.; Matsuki, Y.; Watanabe, E.; Hiramatsu, R.; et al. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab. 2006, 3, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, M.; Anamthathmakula, P.; Sahu, A. Hypothalamic Phosphodiesterase 3B Pathway Mediates Anorectic and Body Weight-Reducing Effects of Insulin in Male Mice. Neuroendocrinology 2017, 104, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Sahu, M.; Anamthathmakula, P.; Sahu, A. Hypothalamic PDE3B deficiency alters body weight and glucose homeostasis in mouse. J. Endocrinol. 2018, 239, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.Z.; Huan, J.-N.; Gupta, S.; Pal, R.; Sahu, A. A phosphatidylinositol 3-kinase–phosphodiesterase 3B–cyclic AMP pathway in hypothalamic action of leptin on feeding. Nat. Neurosci. 2002, 5, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Koshinaka, K.; Sahu, M. PI3K is an upstream regulator of the PDE3B pathway of leptin signaling that may not involve activation of Akt in the rat hypothalamus. J. Neuroendocrinol. 2013, 25, 168–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, T.; O’Hare, J.; Diggs-Andrews, K.; Schweiger, M.; Cheng, B.; Lindtner, C.; Zielinski, E.; Vempati, P.; Su, K.; Dighe, S.; et al. Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metab. 2011, 13, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, A.C.; Filatova, N.; Lindtner, C.; Chi, T.; Degann, S.; Oberlin, D.; Buettner, C. Insulin Receptor Signaling in POMC, but Not AgRP, Neurons Controls Adipose Tissue Insulin Action. Diabetes 2017, 66, 1560–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steculorum, S.M.; Ruud, J.; Karakasilioti, I.; Backes, H.; Ruud, L.E.; Timper, K.; Hess, M.E.; Tsaousidou, E.; Mauer, J.; Vogt, M.C.; et al. AgRP Neurons Control Systemic Insulin Sensitivity via Myostatin Expression in Brown Adipose Tissue. Cell 2016, 165, 125–138. [Google Scholar] [CrossRef]
- Kullmann, S.; Heni, M.; Veit, R.; Scheffler, K.; Machann, J.; Häring, H.-U.; Fritsche, A.; Preissl, H. Selective Insulin Resistance in Homeostatic and Cognitive Control Brain Areas in Overweight and Obese Adults. Diabetes Care 2015, 38, 1044–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiedemann, L.J.; Schmid, S.M.; Hettel, J.; Giesen, K.; Francke, P.; Büchel, C.; Brassen, S. Central insulin modulates food valuation via mesolimbic pathways. Nat. Commun. 2017, 8, 16052. [Google Scholar] [CrossRef] [Green Version]
- Labouèbe, G.; Liu, S.; Dias, C.; Zou, H.; Wong, J.C.Y.; Karunakaran, S.; Clee, S.M.; Phillips, A.G.; Boutrel, B.; Borgland, S.L. Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat. Neurosci. 2013, 16, 300–308. [Google Scholar] [CrossRef]
- Gray, S.M.; Barrett, E.J. Insulin transport into the brain. Am. J. Physiol.-Cell Physiol. 2018, 315, C125–C136. [Google Scholar] [CrossRef]
- Mc Allister, E.; Pacheco-Lopez, G.; Woods, S.C.; Langhans, W. Inconsistencies in the hypophagic action of intracerebroventricular insulin in mice. Physiol. Behav. 2015, 151, 623–628. [Google Scholar] [CrossRef]
- Ramnanan, C.J.; Edgerton, D.S.; Cherrington, A.D. Evidence against a physiologic role for acute changes in CNS insulin action in the rapid regulation of hepatic glucose production. Cell Metab. 2012, 15, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Ott, V.; Lehnert, H.; Staub, J.; Wönne, K.; Born, J.; Hallschmid, M. Central Nervous Insulin Administration Does Not Potentiate the Acute Glucoregulatory Impact of Concurrent Mild Hyperinsulinemia. Diabetes 2015, 64, 760–765. [Google Scholar] [CrossRef]
- Dash, S.; Xiao, C.; Morgantini, C.; Koulajian, K.; Lewis, G.F. Is Insulin Action in the Brain Relevant in Regulating Blood Glucose in Humans? J. Clin. Endocrinol. Metab. 2015, 100, 2525–2531. [Google Scholar] [CrossRef] [Green Version]
- Edgerton, D.S.; Cherrington, A.D. Is Brain Insulin Action Relevant to the Control of Plasma Glucose in Humans? Diabetes 2015, 64, 696–699. [Google Scholar] [CrossRef] [Green Version]
- Hallschmid, M.; Benedict, C.; Schultes, B.; Born, J.; Kern, W. Obese men respond to cognitive but not to catabolic brain insulin signaling. Int. J. Obes. 2008, 32, 275–282. [Google Scholar] [CrossRef]
- Posey, K.A.; Clegg, D.J.; Printz, R.L.; Byun, J.; Morton, G.J.; Vivekanandan-Giri, A.; Pennathur, S.; Baskin, D.G.; Heinecke, J.W.; Woods, S.C.; et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1003–E1012. [Google Scholar] [CrossRef] [Green Version]
- Ono, H.; Pocai, A.; Wang, Y.; Sakoda, H.; Asano, T.; Backer, J.M.; Schwartz, G.J.; Rossetti, L. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. J. Clin. Investig. 2008, 118, 2959–2968. [Google Scholar] [CrossRef] [Green Version]
- Stein, L.J.; Dorsa, D.M.; Baskin, D.G.; Figlewicz, D.P.; Ikeda, H.; Frankmann, S.P.; Greenwood, M.R.; Porte, D.; Woods, S.C. Immunoreactive insulin levels are elevated in the cerebrospinal fluid of genetically obese Zucker rats. Endocrinology 1983, 113, 2299–2301. [Google Scholar] [CrossRef]
- Adam, C.L.; Findlay, P.A.; Aitken, R.P.; Milne, J.S.; Wallace, J.M. In Vivo Changes in Central and Peripheral Insulin Sensitivity in a Large Animal Model of Obesity. Endocrinology 2012, 153, 3147–3157. [Google Scholar] [CrossRef]
- Owen, O.E.; Reichard, G.A.; Boden, G.; Shuman, C. Comparative measurements of glucose, beta-hydroxybutyrate, acetoacetate, and insulin in blood and cerebrospinal fluid during starvation. Metab. Clin. Exp. 1974, 23, 7–14. [Google Scholar] [CrossRef]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.A.; Velloso, L.A. Consumption of a Fat-Rich Diet Activates a Proinflammatory Response and Induces Insulin Resistance in the Hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [CrossRef] [Green Version]
- Banno, R.; Zimmer, D.; Jonghe, B.C.D.; Atienza, M.; Rak, K.; Yang, W.; Bence, K.K. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J. Clin. Investig. 2010, 120, 720–734. [Google Scholar] [CrossRef] [Green Version]
- Loh, K.; Fukushima, A.; Zhang, X.; Galic, S.; Briggs, D.; Enriori, P.J.; Simonds, S.; Wiede, F.; Reichenbach, A.; Hauser, C.; et al. Elevated Hypothalamic TCPTP in Obesity Contributes to Cellular Leptin Resistance. Cell Metab. 2011, 14, 684–699. [Google Scholar] [CrossRef]
- Zhang, Z.-Y.; Dodd, G.T.; Tiganis, T. Protein Tyrosine Phosphatases in Hypothalamic Insulin and Leptin Signaling. Trends Pharmacol. Sci. 2015, 36, 661–674. [Google Scholar] [CrossRef] [Green Version]
- Tsaousidou, E.; Paeger, L.; Belgardt, B.F.; Pal, M.; Wunderlich, C.M.; Brönneke, H.; Collienne, U.; Hampel, B.; Wunderlich, F.T.; Schmidt-Supprian, M.; et al. Distinct Roles for JNK and IKK Activation in Agouti-Related Peptide Neurons in the Development of Obesity and Insulin Resistance. Cell Rep. 2014, 9, 1495–1506. [Google Scholar] [CrossRef] [Green Version]
- Spanswick, D.; Smith, M.A.; Mirshamsi, S.; Routh, V.H.; Ashford, M.L. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat. Neurosci. 2000, 3, 757–758. [Google Scholar] [CrossRef]
- Clegg, D.J.; Gotoh, K.; Kemp, C.; Wortman, M.D.; Benoit, S.C.; Brown, L.M.; D’Alessio, D.; Tso, P.; Seeley, R.J.; Woods, S.C. Consumption of a high-fat diet induces central insulin resistance independent of adiposity. Physiol. Behav. 2011, 103, 10–16. [Google Scholar] [CrossRef]
- Wang, J.; Obici, S.; Morgan, K.; Barzilai, N.; Feng, Z.; Rossetti, L. Overfeeding Rapidly Induces Leptin and Insulin Resistance. Diabetes 2001, 50, 2786–2791. [Google Scholar] [CrossRef] [Green Version]
- Anai, M.; Funaki, M.; Ogihara, T.; Kanda, A.; Onishi, Y.; Sakoda, H.; Inukai, K.; Nawano, M.; Fukushima, Y.; Yazaki, Y.; et al. Enhanced insulin-stimulated activation of phosphatidylinositol 3-kinase in the liver of high-fat-fed rats. Diabetes 1999, 48, 158–169. [Google Scholar] [CrossRef]
- Ono, H. The hypothalamus bridges the gap between physiology and biochemistry in high-fat diet-induced hepatic insulin resistance. Cell Cycle 2009, 8, 2885–2887. [Google Scholar] [CrossRef] [Green Version]
- Jais, A.; Brüning, J.C. Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Investig. 2017, 127, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Romanatto, T.; Cesquini, M.; Amaral, M.E.; Roman, É.A.; Moraes, J.C.; Torsoni, M.A.; Cruz-Neto, A.P.; Velloso, L.A. TNF-α acts in the hypothalamus inhibiting food intake and increasing the respiratory quotient—Effects on leptin and insulin signaling pathways. Peptides 2007, 28, 1050–1058. [Google Scholar] [CrossRef]
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia Dictate the Impact of Saturated Fat Consumption on Hypothalamic Inflammation and Neuronal Function. Cell Rep. 2014, 9, 2124–2138. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Ottaway, N.; Schriever, S.C.; Legutko, B.; García-Cáceres, C.; de la Fuente, E.; Mergen, C.; Bour, S.; Thaler, J.P.; Seeley, R.J.; et al. Hormones and Diet, but Not Body Weight, Control Hypothalamic Microglial Activity. Glia 2014, 62, 17–25. [Google Scholar] [CrossRef]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.L.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated Fatty Acids Produce an Inflammatory Response Predominantly through the Activation of TLR4 Signaling in Hypothalamus: Implications for the Pathogenesis of Obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Kleinridders, A.; Schenten, D.; Könner, A.C.; Belgardt, B.F.; Mauer, J.; Okamura, T.; Wunderlich, F.T.; Medzhitov, R.; Brüning, J.C. MyD88 signaling in the CNS is required for development of fatty acid induced leptin resistance and diet-induced obesity. Cell Metab. 2009, 10, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Won, J.C.; Jang, P.-G.; Namkoong, C.; Koh, E.H.; Kim, S.K.; Park, J.-Y.; Lee, K.-U.; Kim, M.-S. Central Administration of an Endoplasmic Reticulum Stress Inducer Inhibits the Anorexigenic Effects of Leptin and Insulin. Obesity 2009, 17, 1861–1865. [Google Scholar] [CrossRef] [Green Version]
- Contreras, C.; González-García, I.; Martínez-Sánchez, N.; Seoane-Collazo, P.; Jacas, J.; Morgan, D.A.; Serra, D.; Gallego, R.; Gonzalez, F.; Casals, N.; et al. Central Ceramide-Induced Hypothalamic Lipotoxicity and ER Stress Regulate Energy Balance. Cell Rep. 2014, 9, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef]
- Gao, Y.; Bielohuby, M.; Fleming, T.; Grabner, G.F.; Foppen, E.; Bernhard, W.; Guzmán-Ruiz, M.; Layritz, C.; Legutko, B.; Zinser, E.; et al. Dietary sugars, not lipids, drive hypothalamic inflammation. Mol. Metab. 2017, 6, 897–908. [Google Scholar] [CrossRef]
- Obici, S.; Feng, Z.; Morgan, K.; Stein, D.; Karkanias, G.; Rossetti, L. Central Administration of Oleic Acid Inhibits Glucose Production and Food Intake. Diabetes 2002, 51, 271–275. [Google Scholar] [CrossRef]
- Campana, M.; Bellini, L.; Rouch, C.; Rachdi, L.; Coant, N.; Butin, N.; Bandet, C.L.; Philippe, E.; Meneyrol, K.; Kassis, N.; et al. Inhibition of central de novo ceramide synthesis restores insulin signaling in hypothalamus and enhances β-cell function of obese Zucker rats. Mol. Metab. 2018, 8, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Brönneke, H.S.; et al. Obesity-Induced CerS6-Dependent C16:0 Ceramide Production Promotes Weight Gain and Glucose Intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaillard, T.; Roger, M.; Galinier, A.; Guillou, P.; Benani, A.; Leloup, C.; Casteilla, L.; Pénicaud, L.; Lorsignol, A. Hypothalamic Reactive Oxygen Species Are Required for Insulin-Induced Food Intake Inhibition: An NADPH Oxidase–Dependent Mechanism. Diabetes 2009, 58, 1544–1549. [Google Scholar] [CrossRef] [PubMed]
- Storozhevykh, T.P.; Senilova, Y.E.; Persiyantseva, N.A.; Pinelis, V.G.; Pomytkin, I.A. Mitochondrial respiratory chain is involved in insulin-stimulated hydrogen peroxide production and plays an integral role in insulin receptor autophosphorylation in neurons. BMC Neurosci. 2007, 8, 84. [Google Scholar] [CrossRef]
- Drougard, A.; Fournel, A.; Valet, P.; Knauf, C. Impact of hypothalamic reactive oxygen species in the regulation of energy metabolism and food intake. Front. Neurosci 2015, 9, 56. [Google Scholar] [CrossRef]
- Sugiyama, M.; Banno, R.; Mizoguchi, A.; Tominaga, T.; Tsunekawa, T.; Onoue, T.; Hagiwara, D.; Ito, Y.; Morishita, Y.; Iwama, S.; et al. PTP1B deficiency improves hypothalamic insulin sensitivity resulting in the attenuation of AgRP mRNA expression under high-fat diet conditions. Biochem. Biophys. Res. Commun. 2017, 488, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Picardi, P.K.; Calegari, V.C.; de Oliveira Prada, P.; Contin Moraes, J.; Araújo, E.; Gomes Marcondes, M.C.C.; Ueno, M.; Carvalheira, J.B.C.; Velloso, L.A.; Abdalla Saad, M.J. Reduction of Hypothalamic Protein Tyrosine Phosphatase Improves Insulin and Leptin Resistance in Diet-Induced Obese Rats. Endocrinology 2008, 149, 3870–3880. [Google Scholar] [CrossRef] [Green Version]
- Zabolotny, J.M.; Kim, Y.-B.; Welsh, L.A.; Kershaw, E.E.; Neel, B.G.; Kahn, B.B. Protein-tyrosine Phosphatase 1B Expression Is Induced by Inflammation in Vivo. J. Biol. Chem. 2008, 283, 14230–14241. [Google Scholar] [CrossRef] [Green Version]
- Dodd, G.T.; Lee-Young, R.S.; Brüning, J.C.; Tiganis, T. TCPTP Regulates Insulin Signaling in AgRP Neurons to Coordinate Glucose Metabolism with Feeding. Diabetes 2018, 67, 1246–1257. [Google Scholar] [CrossRef]
- Ladyman, S.R.; Grattan, D.R. Region-Specific Suppression of Hypothalamic Responses to Insulin to Adapt to Elevated Maternal Insulin Secretion During Pregnancy. Endocrinology 2017, 158, 4257–4269. [Google Scholar] [CrossRef]
- Pedroso, J.A.B.; Buonfiglio, D.C.; Cardinali, L.I.; Furigo, I.C.; Ramos-Lobo, A.M.; Tirapegui, J.; Elias, C.F.; Donato, J. Inactivation of SOCS3 in leptin receptor-expressing cells protects mice from diet-induced insulin resistance but does not prevent obesity. Mol. Metab. 2014, 3, 608–618. [Google Scholar] [CrossRef]
- Um, S.H.; Frigerio, F.; Watanabe, M.; Picard, F.; Joaquin, M.; Sticker, M.; Fumagalli, S.; Allegrini, P.R.; Kozma, S.C.; Auwerx, J.; et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004, 431, 200–205. [Google Scholar] [CrossRef]
- Hu, F.; Xu, Y.; Liu, F. Hypothalamic roles of mTOR complex I: Integration of nutrient and hormone signals to regulate energy homeostasis. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E994–E1002. [Google Scholar] [CrossRef]
- Caron, A.; Labbé, S.M.; Lanfray, D.; Blanchard, P.-G.; Villot, R.; Roy, C.; Sabatini, D.M.; Richard, D.; Laplante, M. Mediobasal hypothalamic overexpression of DEPTOR protects against high-fat diet-induced obesity. Mol. Metab. 2016, 5, 102–112. [Google Scholar] [CrossRef]
- Cota, D.; Proulx, K.; Smith, K.A.B.; Kozma, S.C.; Thomas, G.; Woods, S.C.; Seeley, R.J. Hypothalamic mTOR signaling regulates food intake. Science 2006, 312, 927–930. [Google Scholar] [CrossRef]
- Blouet, C.; Ono, H.; Schwartz, G.J. Mediobasal hypothalamic p70 S6 kinase 1 modulates the control of energy homeostasis. Cell Metab. 2008, 8, 459–467. [Google Scholar] [CrossRef]
- Smith, M.A.; Katsouri, L.; Irvine, E.E.; Hankir, M.K.; Pedroni, S.M.A.; Voshol, P.J.; Gordon, M.W.; Choudhury, A.I.; Woods, A.; Vidal-Puig, A.; et al. Ribosomal S6K1 in POMC and AgRP Neurons Regulates Glucose Homeostasis but Not Feeding Behavior in Mice. Cell Rep. 2015, 11, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Sakai, G.; Inoue, I.; Suzuki, T.; Sumita, T.; Inukai, K.; Katayama, S.; Awata, T.; Yamada, T.; Asano, T.; Katagiri, H.; et al. Effects of the activations of three major hepatic Akt substrates on glucose metabolism in male mice. Endocrinology 2017, 158, 2659–2671. [Google Scholar] [CrossRef]
- Ayala, J.E.; Bracy, D.P.; McGuinness, O.P.; Wasserman, D.H. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 2006, 55, 390–397. [Google Scholar] [CrossRef]
- Caron, A.; Labbé, S.M.; Mouchiroud, M.; Huard, R.; Richard, D.; Laplante, M. DEPTOR in POMC neurons affects liver metabolism but is dispensable for the regulation of energy balance. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, R1322–R1331. [Google Scholar] [CrossRef] [Green Version]
- Plum, L.; Ma, X.; Hampel, B.; Balthasar, N.; Coppari, R.; Münzberg, H.; Shanabrough, M.; Burdakov, D.; Rother, E.; Janoschek, R.; et al. Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J. Clin. Investig. 2006, 116, 1886–1901. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Bartolome, C.L.; Low, C.S.; Yi, X.; Chien, C.-H.; Wang, P.; Kong, D. Genetic identification of leptin neural circuits in energy and glucose homeostases. Nature 2018, 556, 505–509. [Google Scholar] [CrossRef]
- Van de Wall, E.; Leshan, R.; Xu, A.W.; Balthasar, N.; Coppari, R.; Liu, S.M.; Jo, Y.H.; MacKenzie, R.G.; Allison, D.B.; Dun, N.J.; et al. Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology 2008, 149, 1773–1785. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, J.; Liu, B.; Lv, Z.; Xia, T.; Xiao, F.; Chen, S.; Guo, F. Central Activating Transcription Factor 4 (ATF4) Regulates Hepatic Insulin Resistance in Mice via S6K1 Signaling and the Vagus Nerve. Diabetes 2013, 62, 2230–2239. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.-Y.; Ajoy, R.; Changou, C.A.; Hsieh, Y.-T.; Wang, Y.-K.; Hoffer, B. CCL5/RANTES contributes to hypothalamic insulin signaling for systemic insulin responsiveness through CCR5. Sci. Rep. 2016, 6, 37659. [Google Scholar] [CrossRef] [Green Version]
- Hersom, M.; Helms, H.C.; Schmalz, C.; Pedersen, T.Å.; Buckley, S.T.; Brodin, B. The insulin receptor is expressed and functional in cultured blood-brain barrier endothelial cells but does not mediate insulin entry from blood to brain. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E531–E542. [Google Scholar] [CrossRef]
- Molnár, G.; Faragó, N.; Kocsis, Á.K.; Rózsa, M.; Lovas, S.; Boldog, E.; Báldi, R.; Csajbók, É.; Gardi, J.; Puskás, L.G.; et al. GABAergic Neurogliaform Cells Represent Local Sources of Insulin in the Cerebral Cortex. J. Neurosci. 2014, 34, 1133–1137. [Google Scholar] [CrossRef] [Green Version]
- Begg, D.P. Chapter Eight—Insulin Transport into the Brain and Cerebrospinal Fluid. In Vitamins & Hormones; Litwack, G., Ed.; Hormones and Transport Systems; Academic Press: Cambridge, MA, USA, 2015; Volume 98, pp. 229–248. [Google Scholar]
- Kaiyala, K.J.; Prigeon, R.L.; Kahn, S.E.; Woods, S.C.; Schwartz, M.W. Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes 2000, 49, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Urayama, A.; Banks, W.A. Starvation and triglycerides reverse the obesity-induced impairment of insulin transport at the blood-brain barrier. Endocrinology 2008, 149, 3592–3597. [Google Scholar] [CrossRef]
- Heni, M.; Schöpfer, P.; Peter, A.; Sartorius, T.; Fritsche, A.; Synofzik, M.; Häring, H.-U.; Maetzler, W.; Hennige, A.M. Evidence for altered transport of insulin across the blood–brain barrier in insulin-resistant humans. Acta Diabetol 2014, 51, 679–681. [Google Scholar] [CrossRef]
- Rahman, M.H.; Bhusal, A.; Lee, W.-H.; Lee, I.-K.; Suk, K. Hypothalamic inflammation and malfunctioning glia in the pathophysiology of obesity and diabetes: Translational significance. Biochem. Pharmacol. 2018, 153, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Argente-Arizón, P.; Guerra-Cantera, S.; Garcia-Segura, L.M.; Argente, J.; Chowen, J.A. Glial cells and energy balance. J. Mol. Endocrinol. 2017, 58, R59–R71. [Google Scholar] [CrossRef] [PubMed]
- Könner, A.C.; Janoschek, R.; Plum, L.; Jordan, S.D.; Rother, E.; Ma, X.; Xu, C.; Enriori, P.; Hampel, B.; Barsh, G.S.; et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007, 5, 438–449. [Google Scholar] [CrossRef]
- Dodd, G.T.; Michael, N.J.; Lee-Young, R.S.; Mangiafico, S.P.; Pryor, J.T.; Munder, A.C.; Simonds, S.E.; Brüning, J.C.; Zhang, Z.-Y.; Cowley, M.A.; et al. Insulin regulates POMC neuronal plasticity to control glucose metabolism. Elife 2018, 7, e38704. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.; Zhang, L.; Brandon, A.; Wang, Q.; Begg, D.; Qi, Y.; Fu, M.; Kulkarni, R.; Teo, J.; Baldock, P.; et al. Insulin controls food intake and energy balance via NPY neurons. Mol. Metab. 2017, 6, 574–584. [Google Scholar] [CrossRef]
- Choi, S.J.; Kim, F.; Schwartz, M.W.; Wisse, B.E. Cultured hypothalamic neurons are resistant to inflammation and insulin resistance induced by saturated fatty acids. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1122–E1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaler, J.P.; Yi, C.-X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef]
- André, C.; Guzman-Quevedo, O.; Rey, C.; Rémus-Borel, J.; Clark, S.; Castellanos-Jankiewicz, A.; Ladeveze, E.; Leste-Lasserre, T.; Nadjar, A.; Abrous, D.N.; et al. Inhibiting Microglia Expansion Prevents Diet-Induced Hypothalamic and Peripheral Inflammation. Diabetes 2017, 66, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Berkseth, K.E.; Guyenet, S.J.; Melhorn, S.J.; Lee, D.; Thaler, J.P.; Schur, E.A.; Schwartz, M.W. Hypothalamic Gliosis Associated with High-Fat Diet Feeding Is Reversible in Mice: A Combined Immunohistochemical and Magnetic Resonance Imaging Study. Endocrinology 2014, 155, 2858–2867. [Google Scholar] [CrossRef]
- Buckman, L.B.; Thompson, M.M.; Lippert, R.N.; Blackwell, T.S.; Yull, F.E.; Ellacott, K.L.J. Evidence for a novel functional role of astrocytes in the acute homeostatic response to high-fat diet intake in mice. Mol. Metab. 2015, 4, 58–63. [Google Scholar] [CrossRef]
- Zhang, Y.; Reichel, J.M.; Han, C.; Zuniga-Hertz, J.P.; Cai, D. Astrocytic process plasticity and IKKβ/NF-κB in central control of blood glucose, blood pressure and body weight. Cell Metab. 2017, 25, 1091–1102.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, A.; Prevot, V. When Size Matters: How Astrocytic Processes Shape Metabolism. Cell Metab. 2017, 25, 995–996. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.-X.; Gericke, M.; Krüger, M.; Alkemade, A.; Kabra, D.G.; Hanske, S.; Filosa, J.; Pfluger, P.; Bingham, N.; Woods, S.C.; et al. High calorie diet triggers hypothalamic angiopathy. Mol. Metab. 2012, 1, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kim, H.J.; Lee, Y.-S.; Kang, G.M.; Lim, H.S.; Lee, S.; Song, D.K.; Kwon, O.; Hwang, I.; Son, M.; et al. Hypothalamic Macrophage Inducible Nitric Oxide Synthase Mediates Obesity-Associated Hypothalamic Inflammation. Cell Rep. 2018, 25, 934–946.e5. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ono, H. Molecular Mechanisms of Hypothalamic Insulin Resistance. Int. J. Mol. Sci. 2019, 20, 1317. https://doi.org/10.3390/ijms20061317
Ono H. Molecular Mechanisms of Hypothalamic Insulin Resistance. International Journal of Molecular Sciences. 2019; 20(6):1317. https://doi.org/10.3390/ijms20061317
Chicago/Turabian StyleOno, Hiraku. 2019. "Molecular Mechanisms of Hypothalamic Insulin Resistance" International Journal of Molecular Sciences 20, no. 6: 1317. https://doi.org/10.3390/ijms20061317
APA StyleOno, H. (2019). Molecular Mechanisms of Hypothalamic Insulin Resistance. International Journal of Molecular Sciences, 20(6), 1317. https://doi.org/10.3390/ijms20061317