Confinement Effect of Micro- and Mesoporous Materials on the Spectroscopy and Dynamics of a Stilbene Derivative Dye



Abstract
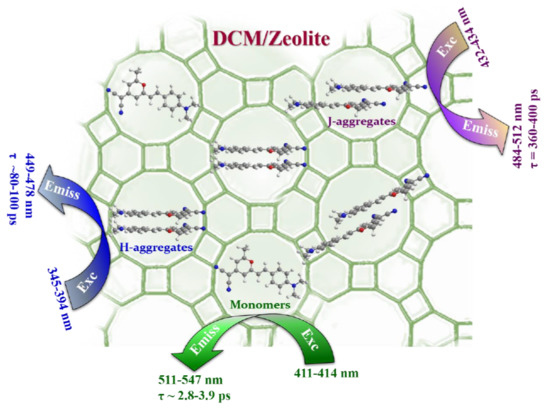
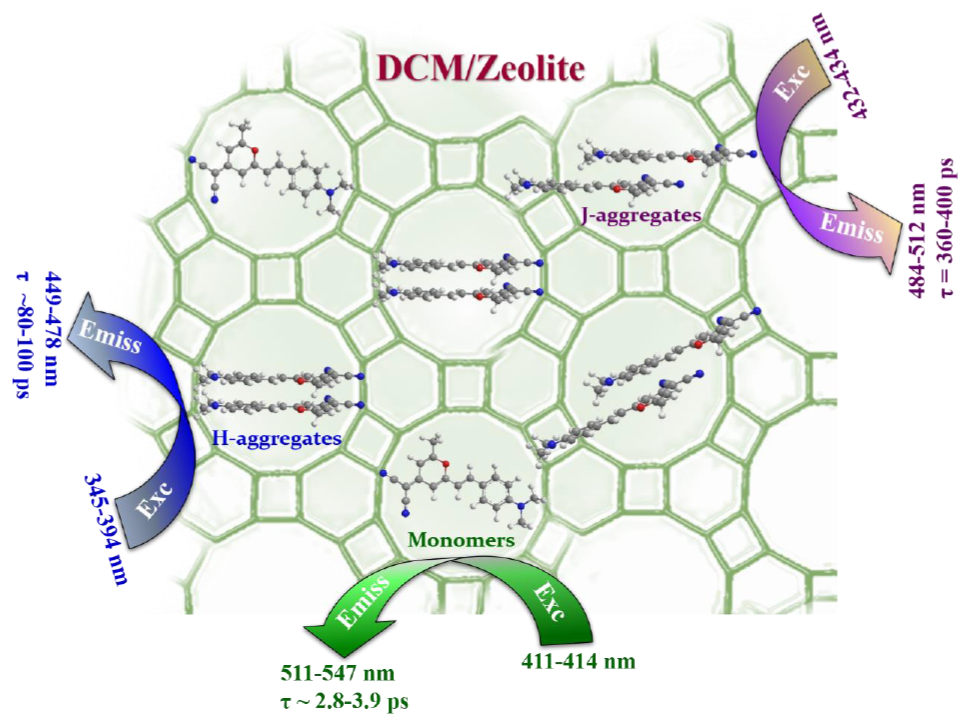
:
1. Introduction
2. Results and Discussion
2.1. Steady-State Observation
2.1.1. Dichloromethane Solution
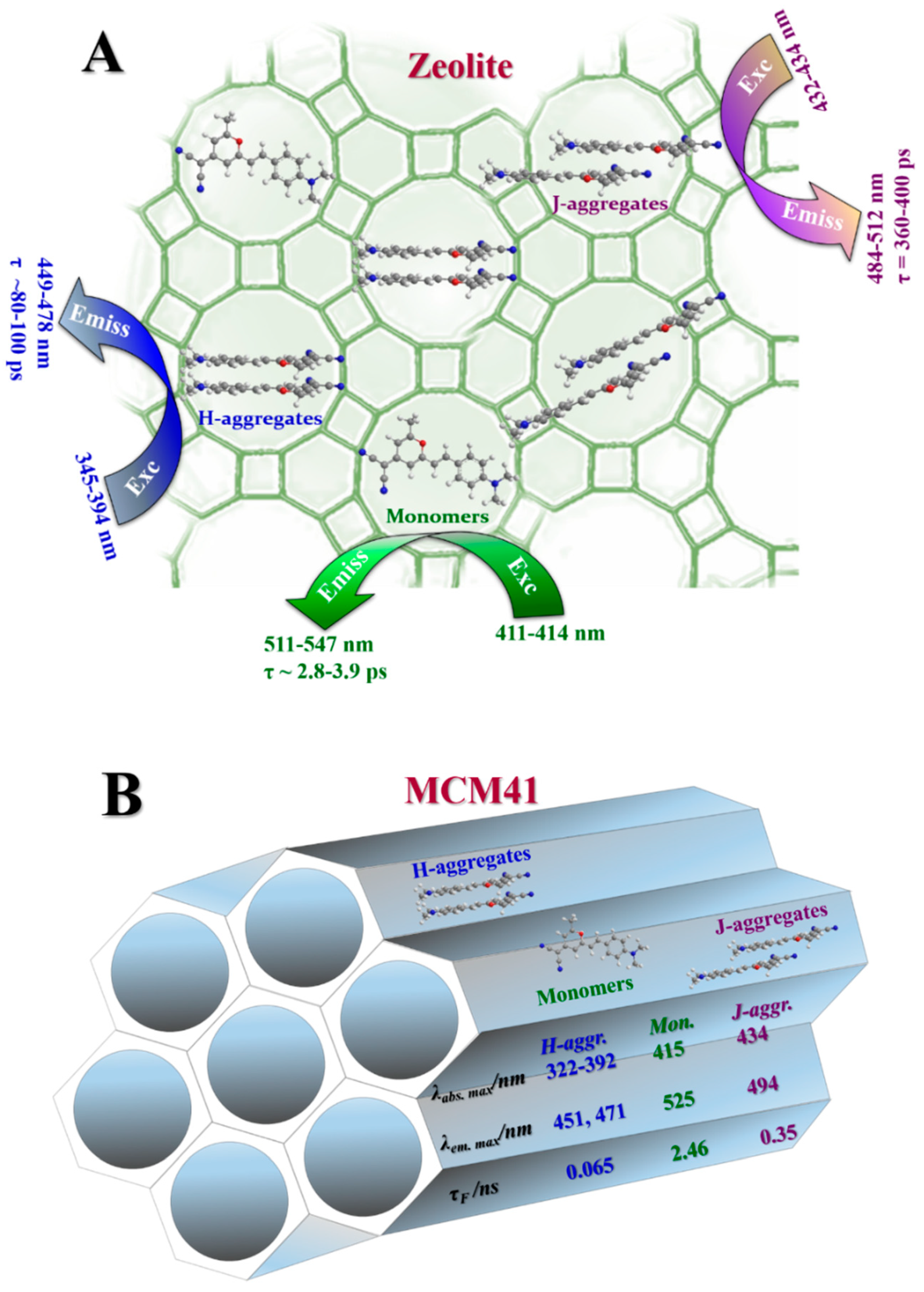
2.1.2. Complexes of DCM with HY, NaX, NaY Zeolites, and MCM-41
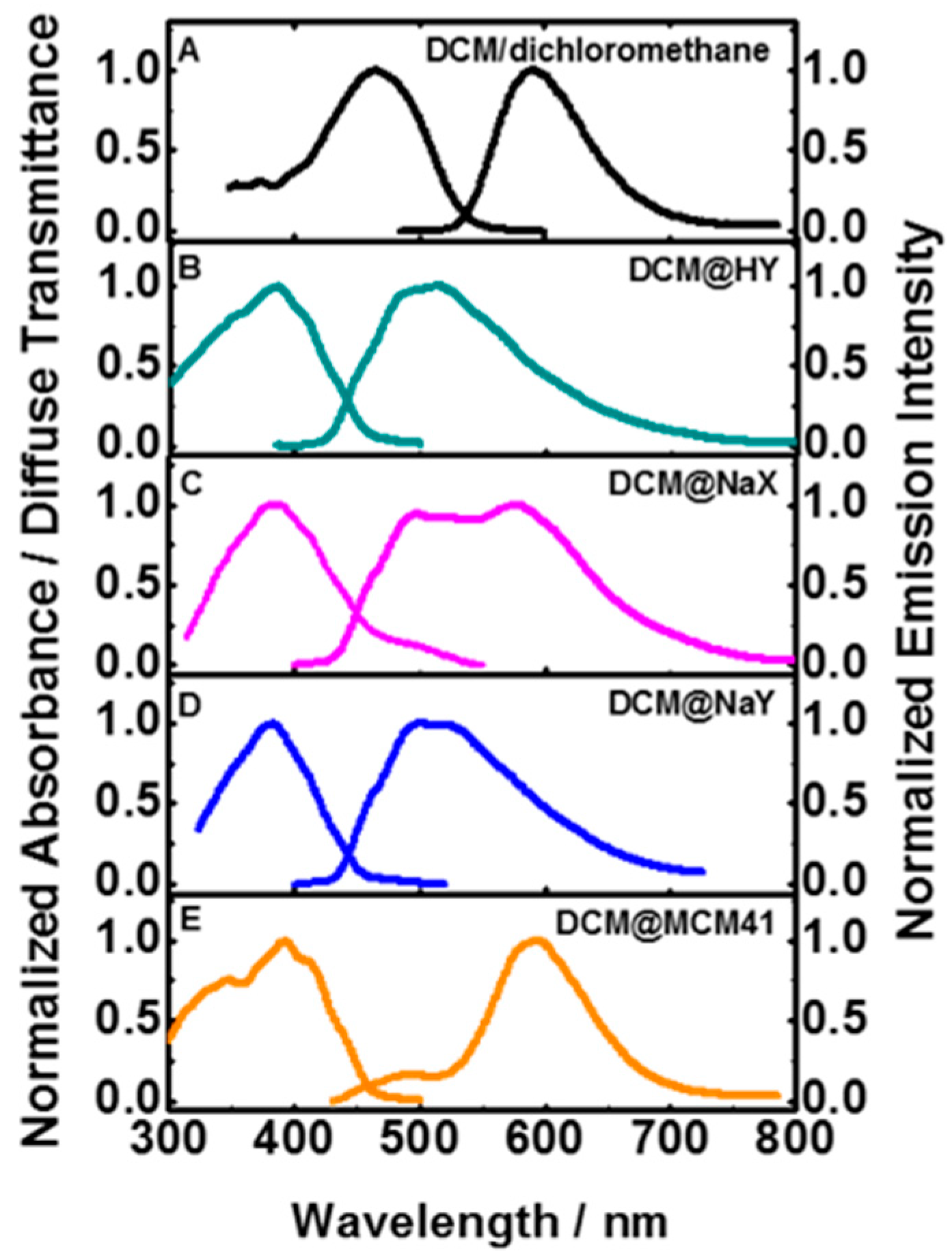
Diffuse Transmittance Spectra
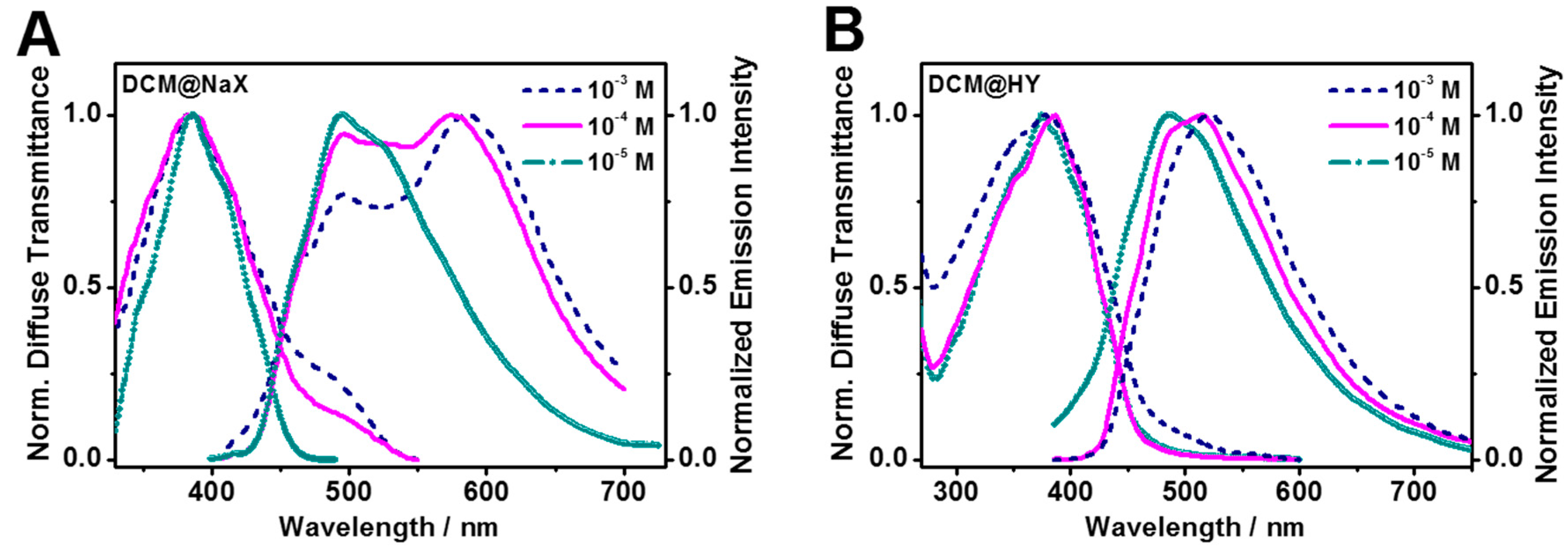
Concentration Effect on the DT Spectra of the DCM@zeolite Complexes
Deconvolution Analysis of the DT Spectra of DCM Interacting with the Studied Microporous and Mesoporous Materials
Differences in DCM Loading Capability of the Studied Microporous and Mesoporous Materials
Steady-State Emission Spectra
Concentration Effect on the Emission Spectra of the DCM@zeolite Complexes
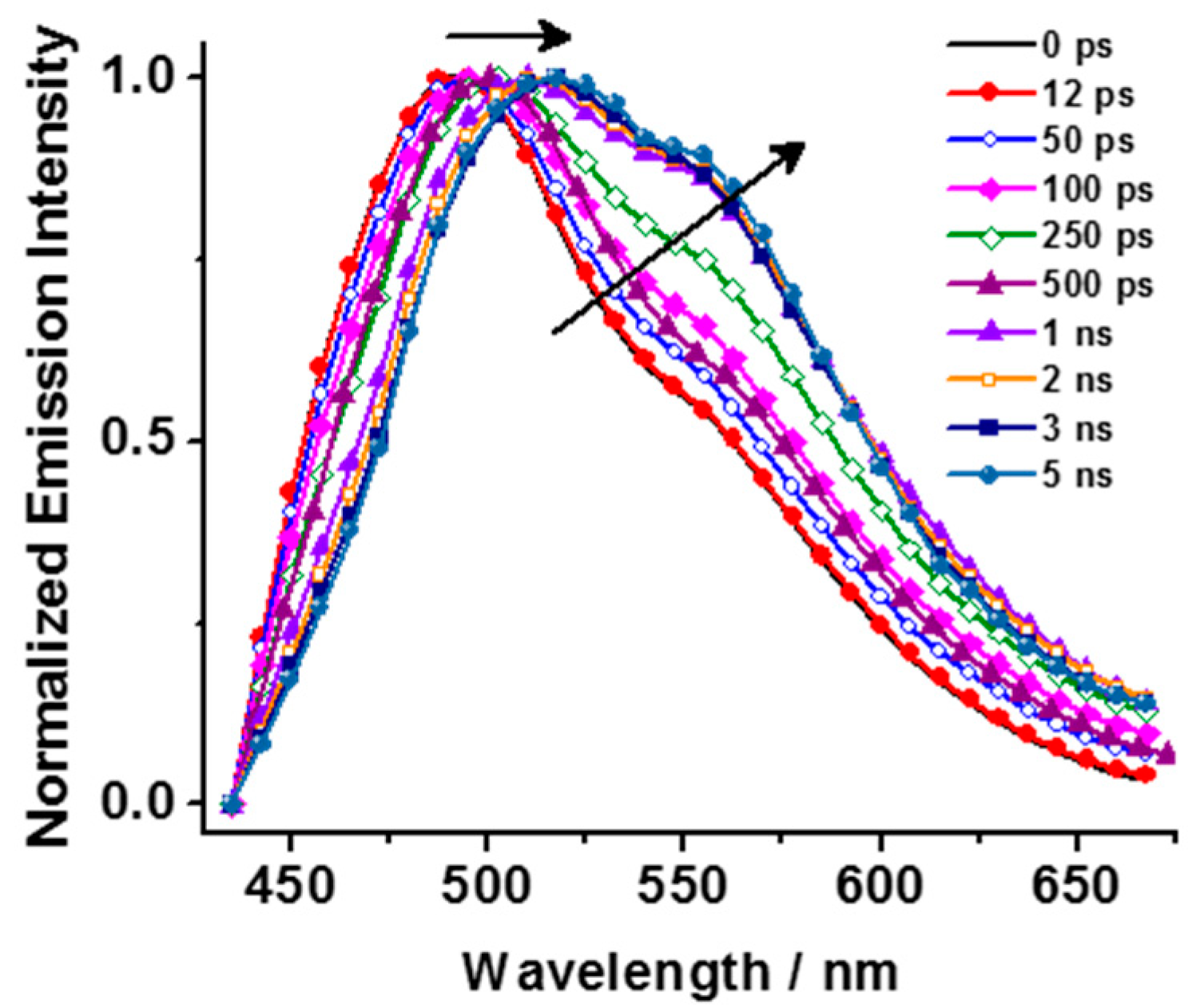
2.2. Time-Resolved Emission Measurements—Emission Decays of DCM Interacting with HY, NaX, and NaY Zeolites in Dichloromethane Suspensions
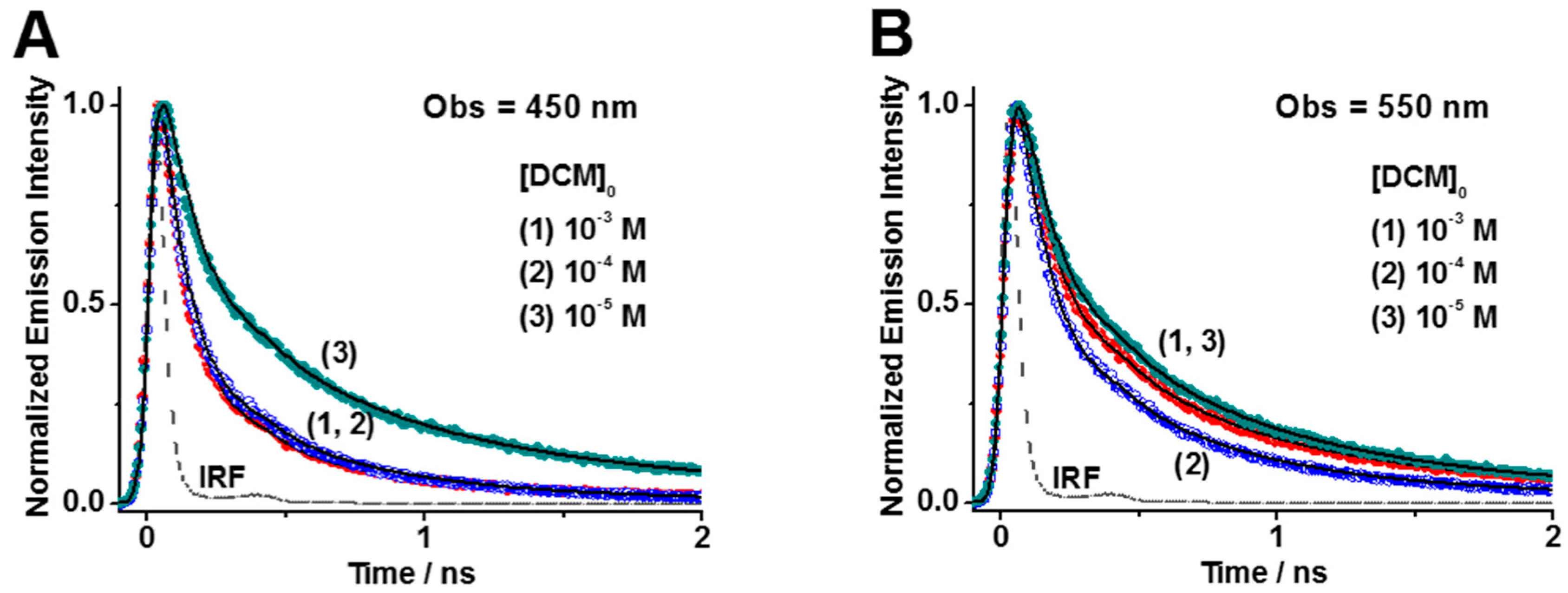
2.2.1. Concentration Effect on the Photobehavior of DCM@HY
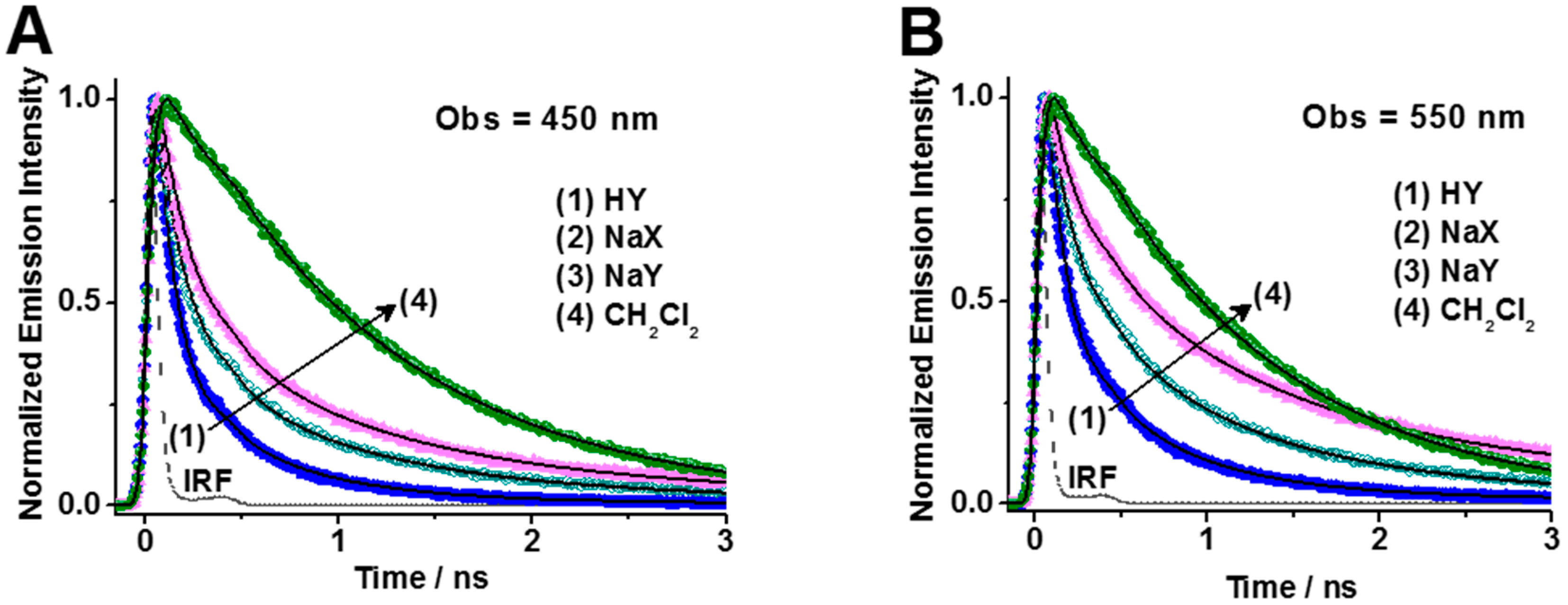
2.2.2. Photobehavior of DCM within HY, NaX, and NaY Zeolites
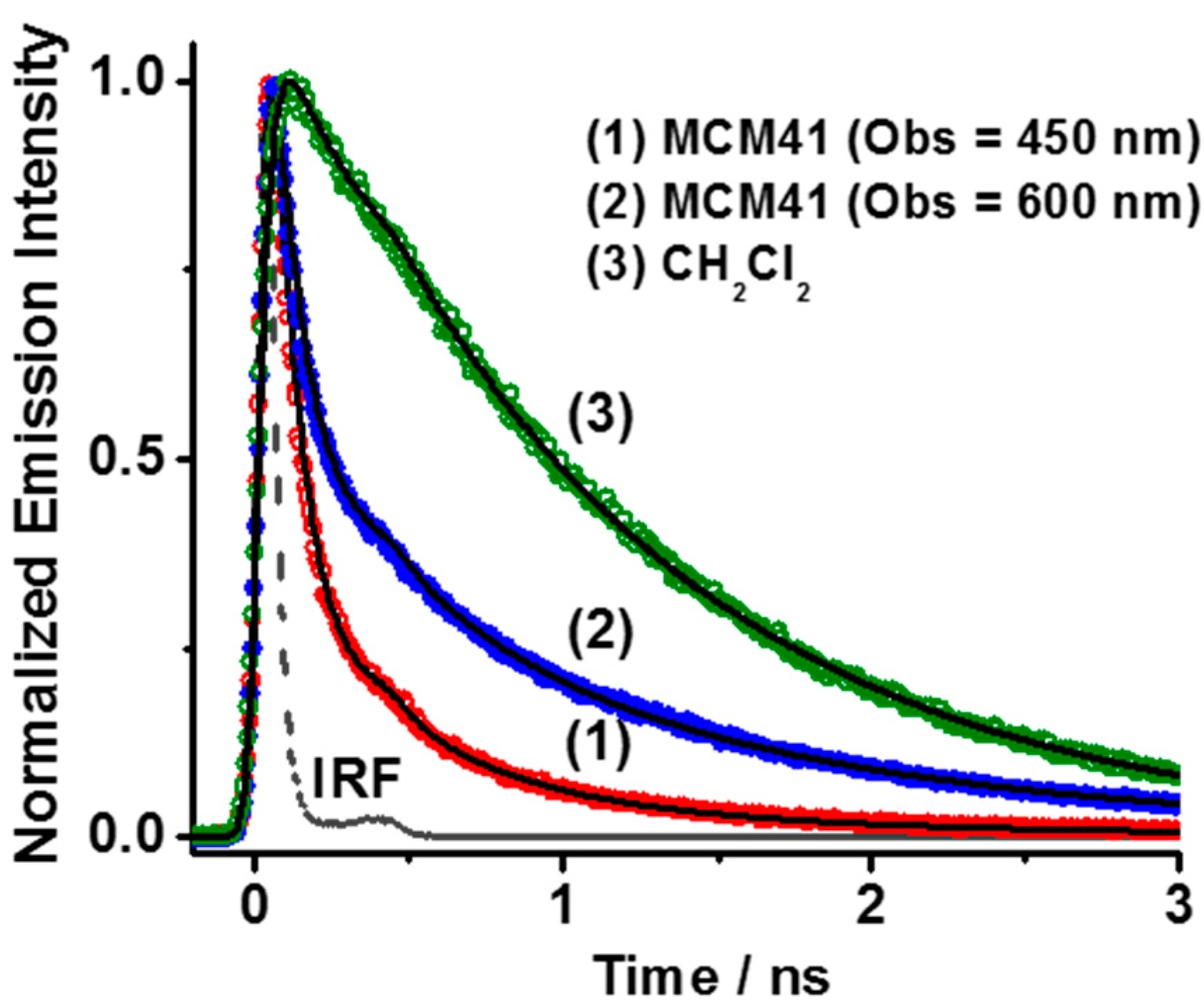
2.2.3. Emission Decays of DCM Interacting with MCM-41 in Dichloromethane Suspensions.
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DCM | trans-4-(dicyanomethylene)-2-methyl-6-(4-dimethylaminostyryl)-4H-pyran |
| CT | charge-transfer |
| HBA-4NP | E)-2-(2-hydroxybenzyliden)amino-4-nitrophenol |
| D | donor |
| A | acceptor |
| ICT | intramolecular charge-transfer |
| LE | locally excited |
| HSA | human serum albumin |
| PVK | polyvinyl carbazole |
| TICT | twisted intramolecular charge-transfer |
| Zr-NDC | Zr-naphthalene dicarboxylic acid |
| MOF | metal organic framework |
| DFT | density functional theory |
| CASSCF | complete active space self-consistent-field |
| DT | diffuse transmittance |
| τi | time constant |
| ai | pre-exponential factor |
| τav | average lifetime |
| TRES | time-resolved emission spectra |
| TCSPC | time-correlated single-photon-counting |
| IRF | instrument response function |
| Ai | initial absorbance |
| As | supernatant absorbance |
| ε | molar extinction coefficient |
| l | optical path length |
| V | volume |
| NA | Avogadro’s number |
References
- Alarcos, N.; Cohen, B.; Ziółek, M.; Douhal, A. Photochemistry and Photophysics in Silica-Based Materials: Ultrafast and Single Molecule Spectroscopy Observation. Chem. Rev. 2017, 117, 13639–13720. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Yu, H.; Kwon, O.-H.; Jang, D.-J. Photo-Induced Proton-Transfer Cycle of 2-Naphthol in Faujasite Zeolitic Nanocavities. Phys. Chem. Chem. Phys. 2008, 10, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Alarcos, N.; Cohen, B.; Douhal, A. A Slowing Down of Proton Motion from HPTS to Water Adsorbed on the MCM-41 Surface. Phys. Chem. Chem. Phys. 2016, 18, 2658–2671. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Yu, H.; Park, J.; Jang, D.-J. Excited-State Prototropic Equilibrium Dynamics of 6-Hydroxyquinoline Encapsulated in Microporous Catalytic Faujasite Zeolites. Chem. Eur. J. 2010, 16, 12609–12615. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Amino, Y.; Shima, K.; Suzuki, S.; Yamashita, T.; Teramae, N. Local Environments of Coumarin Dyes within Mesostructured Silica-Surfactant Nanocomposites. J. Phys. Chem. B 2006, 110, 3910–3916. [Google Scholar] [CrossRef] [PubMed]
- Ziółek, M.; Martín, C.; Navarro, M.T.; Garcia, H.; Douhal, A. Confined Photodynamics of an Organic Dye for Solar Cells Encapsulated in Titanium-Doped Mesoporous Molecular Materials. J. Phys. Chem. C 2011, 115, 8858–8867. [Google Scholar] [CrossRef]
- Keirstead, A.E.; Schepp, N.P.; Cozens, F.L. Influence of the Alkali Metal Cation on the Distance of Electron Migration in Zeolite Y: A Nanosecond Laser Photolysis Study. J. Phys. Chem. C 2007, 111, 14247–14252. [Google Scholar] [CrossRef]
- Lim, H.; Choi, S.-E.; Cheong, H.; Lee, J.S. Energy Transfer in Dye Molecule-Containing Zeolite Monolayers. Microporous Mesoporous Mater. 2014, 192, 89–94. [Google Scholar] [CrossRef]
- Hu, D.D.; Lin, J.; Zhang, Q.; Lu, J.N.; Wang, X.Y.; Wang, Y.W.; Bu, F.; Ding, L.F.; Wang, L.; Wu, T. Multi-Step Host-Guest Energy Transfer between Inorganic Chalcogenide-Based Semiconductor Zeolite Material and Organic Dye Molecules. Chem. Mater. 2015, 27, 4099–4104. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Chen, F.; Zhang, J.; Anpo, M. Manipulating Energy Transfer Processes between Rhodamine 6G and Rhodamine B in Different Mesoporous Hosts. J. Phys. Chem. C 2007, 111, 5541–5548. [Google Scholar] [CrossRef]
- Alarcos, N.; Organero, J.A.; Sánchez, F.; Douhal, A. Exploring the Photobehavior of Nanocaged Monomers and H- and J-aggregates of a Proton-Transfer Dye within NaX and NaY Zeolites. J. Phys. Chem. C 2014, 118, 8217–8226. [Google Scholar] [CrossRef]
- Alarcos, N.; Sánchez, F.; Douhal, A. Spectroscopy and Relaxation Dynamics of Salicylideneaniline Derivative Aggregates Encapsulated in MCM41 and SBA15 Pores. Microporous Mesoporous Mater. 2016, 226, 34–43. [Google Scholar] [CrossRef]
- Grabowski, Z.R.; Rotkiewicz, K.; Rettig, W. Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures. Chem. Rev. 2003, 103, 3899–4032. [Google Scholar] [CrossRef]
- Karunakaran, V.; Das, S. Direct Observation of Cascade of Photoinduced Ultrafast Intramolecular Charge Transfer Dynamics in Diphenyl Acetylene Derivatives: Via Solvation and Intramolecular Relaxation. J. Phys. Chem. B 2016, 120, 7016–7023. [Google Scholar] [CrossRef] [PubMed]
- Wiedbrauk, S.; Maerz, B.; Samoylova, E.; Reiner, A.; Trommer, F.; Mayer, P.; Zinth, W.; Dube, H. Twisted Hemithioindigo Photoswitches: Solvent Polarity Determines the Type of Light-Induced Rotations. J. Am. Chem. Soc. 2016, 138, 12219–12227. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.; Wang, S.; Organero, J.A.; Teruel, L.; Garcia, H.; Douhal, A. Femtosecond Dynamics within Nanotubes and Nanocavities of Mesoporous and Zeolite Materials. J. Phys. Chem. C 2009, 113, 11614–11622. [Google Scholar] [CrossRef]
- Gil, M.; Organero, J.A.; Peris, E.; Garcia, H.; Douhal, A. Confinement Effect of Nanocages and Nanotubes of Mesoporous Materials on the Keto Forms Photodynamics of Sudan I. Chem. Phys. Lett. 2009, 474, 325–330. [Google Scholar] [CrossRef]
- Martin, C.; Cohen, B.; Navarro, M.T.; Corma, A.; Douhal, A. Unraveling the Ultrafast Behavior of Nile Red Interacting with Aluminum and Titanium Co-Doped MCM41 Materials. Phys. Chem. Chem. Phys. 2016, 18, 2152–2163. [Google Scholar] [CrossRef]
- Martín, C.; Piatkowski, P.; Cohen, B.; Gil, M.; Navarro, M.T.; Corma, A.; Douhal, A. Ultrafast Dynamics of Nile Red Interacting with Metal Doped Mesoporous Materials. J. Phys. Chem. C 2015, 119, 13283–13296. [Google Scholar] [CrossRef]
- Freidzon, A.Y.; Safonov, A.A.; Bagaturyants, A.A.; Alfimov, M.V. Solvatofluorochromism and Twisted Intramolecular Charge-Transfer State of the Nile Red Dye. Int. J. Quantum Chem. 2012, 112, 3059–3067. [Google Scholar] [CrossRef]
- Sarkar, N.; Das, K.; Nath, D.N.; Bhattacharyya, K. Twisted Charge Transfer Process of Nile Red in Homogeneous Solution and in Faujasite Zeolite. Langmuir 1994, 10, 326–329. [Google Scholar] [CrossRef]
- Sackett, D.L.; Wolff, J. Nile Red as a Polarity-Sensitive Fluorescent Probe of Hydrophobic Protein Surfaces. Anal. Biochem. 1987, 167, 228–234. [Google Scholar] [CrossRef]
- Dutta, A.K.; Kamada, K.; Ohta, K. Spectroscopic Studies of Nile Red in Organic Solvents and Polymers. J. Photochem. Photobiol. A 1996, 93, 57–64. [Google Scholar] [CrossRef]
- Alarcos, N.; Cohen, B.; Douhal, A. Photodynamics of a Proton-Transfer Dye in Solutions and Confined within NaX and NaY Zeolites. J. Phys. Chem. C 2014, 118, 19431–19443. [Google Scholar] [CrossRef]
- Meyer, M.; Mialocq, J.C. Ground State and Singlet Excited State of Laser Dye DCM: Dipole Moments and Solvent Induced Spectral Shifts. Opt. Commun. 1987, 64, 264–268. [Google Scholar] [CrossRef]
- Meyer, M.; Mialocq, J.C.; Perly, B. Photoinduced Intramolecular Charge Transfer and Trans-Cis Isomerization of the DCM Styrene Dye: Picosecond and Nanosecond Laser Spectroscopy, High-Performance Liquid Chromatography, and Nuclear Magnetic Resonance Studies. J. Phys. Chem. 1990, 94, 98–104. [Google Scholar] [CrossRef]
- Mialocq, J.C.; Armand, X.; Marguet, S. A New Sensitive Chemical Actinometer for Time-Resolved and Continuous Photochemistry: The DCM Styrene Dye. J. Photochem. Photobiol. A 1993, 69, 351–356. [Google Scholar] [CrossRef]
- Martin, M.M.; Plaza, P.; Meyer, Y.H. Ultrafast Intramolecular Charge Transfer in the Merocyanine Dye DCM. Chem. Phys. 1995, 192, 367–377. [Google Scholar] [CrossRef]
- Gustavsson, T.; Baldacchino, G.; Mialocq, J.C.; Pommeret, S. A Femtosecond Fluorescence Up-Conversion Study of the Dynamic Stokes Shift of the DCM Dye Molecule in Polar and Non-Polar Solvents. Chem. Phys. Lett. 1995, 236, 587–594. [Google Scholar] [CrossRef]
- Pommeret, S.; Gustavsson, T.; Naskrecki, R.; Baldacchino, G.; Mialocq, J.C. Femtosecond Absorption and Emission Spectroscopy of the DCM Laser Dye. J. Mol. Liq. 1995, 64, 101–112. [Google Scholar] [CrossRef]
- Zhang, H.; Jonkman, A.M.; Vandermeulen, P.; Glasbeek, M. Femtosecond Studies of Charge Separation in Phot-Excited DCM in Liquid Solution. Chem. Phys. Lett. 1994, 224, 551–556. [Google Scholar] [CrossRef]
- Maciejewski, A.; Naskrecki, R.; Lorenc, M.; Ziolek, M.; Karolczak, J.; Kubicki, J.; Matysiak, M.; Szymanski, M. Transient Absorption Experimental Set-Up with Femtosecond Time Resolution. Femto- and Picosecond Study of DCM Molecule in Cyclohexane and Methanol Solution. J. Mol. Struct. 2000, 555, 1–13. [Google Scholar] [CrossRef]
- Van Tassle, A.J.; Prantil, M.A.; Fleming, G.R. Investigation of the Excited State Structure of DCM via Ultrafast Electronic Pump/Vibrational Probe. J. Phys. Chem. B 2006, 110, 18989–18995. [Google Scholar] [CrossRef] [PubMed]
- Marguet, S.; Mialocq, J.C.; Millie, P.; Berthier, G.; Momicchioli, F. Intramolecular Charge Transfer and Trans-Cis Isomerization of the DCM Styrene Dye in Polar Solvents. A CS INDO MRCI Study. Chem. Phys. 1992, 160, 265–279. [Google Scholar] [CrossRef]
- Petsalakis, I.D.; Georgiadou, D.G.; Vasilopoulou, M.; Pistolis, G.; Dimotikali, D.; Argitis, P.; Theodorakopoulos, G. Theoretical Investigation on the Effect of Protonation on the Absorption and Emission Spectra of Two Amine-Group-Bearing, Red “Push-Pull” Emitters, 4-Dimethylamino-4’-nitrostilbene and 4-(Dicyanomethylene)-2-methyl-6-p-(dimethylamino) styryl-4H-pyran, by DFT and TDDFT Calculations. J. Phys. Chem. A 2010, 114, 5580–5587. [Google Scholar] [PubMed]
- Pal, S.K.; Sukul, D.; Mandal, D.; Sen, S.; Bhattacharyya, K. Solvation Dynamics of DCM in Micelles. Chem. Phys. Lett. 2000, 327, 91–96. [Google Scholar] [CrossRef]
- Mandal, D.; Sen, S.; Bhattacharyya, K.; Tahara, T. Femtosecond Study of Solvation Dynamics of DCM in Micelles. Chem. Phys. Lett. 2002, 359, 77–82. [Google Scholar] [CrossRef]
- Pal, S.K.; Sukul, D.; Mandal, D.; Bhattacharyya, K. Solvation Dynamics of DCM in Lipid. J. Phys. Chem. B 2000, 104, 4529–4531. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.K.; Sukul, D.; Mandal, D.; Sen, S.; Bhattacharyya, K. Solvation Dynamics of DCM in Dipalmitoyl Phosphatidylcholine Lipid. Tetrahedron 2000, 56, 6999–7002. [Google Scholar] [CrossRef]
- Pal, S.K.; Mandal, D.; Sukul, D.; Bhattacharyya, K. Solvation Dynamics of 4-(Dicyanomethylene)-2-methyl-6-(p-dimethylaminostyryl)-4H-pyran (DCM) in a Microemulsion. Chem. Phys. Lett. 1999, 312, 178–184. [Google Scholar] [CrossRef]
- Pal, S.K.; Mandal, D.; Sukul, D.; Sen, S.; Bhattacharyya, K. Solvation Dynamics of DCM in Human Serum Albumin. J. Phys. Chem. B 2001, 105, 1438–1441. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Wang, L.; Liu, Y.; Ning, Y.; Zhao, J.; Liu, X.; Wu, S.; He, X.; Lin, J.; Wang, L.; et al. Lasing Behavior in DCM-doped PVK Microcavity. Synth. Met. 2000, 111–112, 563–565. [Google Scholar] [CrossRef]
- Halder, A.; Sen, P.; Burman, A.D.; Bhattacharyya, K. Solvation Dynamics of DCM in a Polypeptide-Surfactant Aggregate: Gelatin-Sodium Dodecyl Sulfate. Langmuir 2004, 20, 653–657. [Google Scholar] [CrossRef]
- Guo, H.; Zhang, X.; Aydin, M.; Xu, W.; Zhu, H.-R.; Akins, D.L. Spectroscopy and Dynamics of DCM Encapsulated in MCM-41 and Y Zeolite Mesoporous Materials. J. Mol. Struct. 2004, 689, 153–158. [Google Scholar] [CrossRef]
- Gutiérrez, M.; Sánchez, F.; Douhal, A. Efficient Multicolor and White Light Emission from Zr-Based MOF Composites: Spectral and Dynamic Properties. J. Mater. Chem. C 2015, 3, 11300–11310. [Google Scholar] [CrossRef]
- Pomogaev, V.A.; Svetlichnyi, V.A.; Pomogaev, A.V.; Svetlichnaya, N.N.; Kopylova, T.N. Theoretic and Experimental Study of Photoprocesses in Substituted 4-Dicyanomethylene-4H-pyrans. High Energy Chem. 2005, 39, 403–407. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, R.; Cao, Z.; Zhang, Q. Intramolecular Charge Transfer and Photoisomerization of the DCM Styrene Dye: A Theoretical Study. J. Theor. Comput. Chem. 2008, 7, 719–736. [Google Scholar] [CrossRef]
- Su, F.; Zhao, X.S.; Lv, L.; Zhou, Z. Synthesis and characterization of microporous carbons templated by ammonium-form zeolite Y. Carbon 2004, 42, 2821–2831. [Google Scholar] [CrossRef]
- Laborde-Boutet, C.; Joly, G.; Nicolaos, A.; Thomas, M.; Magnoux, P. Selectivity of Thiophene/Toluene Competitive Adsorptions onto NaY and NaX Zeolites. Ind. Eng. Chem. Res. 2006, 45, 6758–6764. [Google Scholar] [CrossRef]
- Busby, M.; Blum, C.; Tibben, M.; Fibikar, S.; Calzaferri, G.; Subramaniam, V.; De Cola, L. Time, Space, and Spectrally Resolved Studies on J-Aggregate Interactions in Zeolite L Nanochannels. J. Am. Chem. Soc. 2008, 130, 10970–10976. [Google Scholar] [CrossRef] [PubMed]
- Nüesch, F.; Moser, J.E.; Shklover, V.; Grätzel, M. Merocyanine Aggregation in Mesoporous Networks. J. Am. Chem. Soc. 1996, 118, 5420–5431. [Google Scholar] [CrossRef]
- Cheng, K.A.W.Y.; Schepp, N.P.; Cozens, F.L. Ultrafast Dynamics of Pyrene Excimer Formation in Y Zeolites. J. Phys. Chem. A 2004, 108, 7132–7134. [Google Scholar] [CrossRef]
- Cheng, K.A.W.Y.; Schepp, N.P.; Cozens, F.L. Resolution of Ultrafast Pyrene Excimer Emission Rise Times in Zeolites X and Y. Photochem. Photobiol. 2006, 82, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Behera, R.K.; Behera, P.K.; Mishra, B.K.; Behera, G.B. Cyanines During the 1990s: A Review. Chem. Rev. 2000, 100, 1973–2011. [Google Scholar] [CrossRef]
- Alarcos, N.; Sánchez, F.; Douhal, A. Confinement Effect on Ultrafast Events of a Salicylideneaniline Derivative within Mesoporous Materials. Microporous Mesoporous Mater. 2017, 248, 54–61. [Google Scholar] [CrossRef]
- Hestand, N.J.; Spano, F.C. Expanded Theory of H- and J-Molecular Aggregates: The Effects of Vibronic Coupling and Intermolecular Charge Transfer. Chem. Rev. 2018, 118, 7069–7163. [Google Scholar] [CrossRef]
- Kasha, M.; Rawls, H.R.; Ashraf El-Bayoumi, M. The Exciton Model in Molecular Spectroscopy. Pure Appl. Chem. 1965, 11, 371–392. [Google Scholar] [CrossRef]
- Eisfeld, A.; Briggs, J.S. The J- and H-Bands of Organic Dye Aggregates. Chem. Phys. 2006, 324, 376–384. [Google Scholar] [CrossRef]
- Davydov, A. Theory of Molecular Excitons; Springer: Boston, MA, USA, 2013. [Google Scholar]
- Kobayashi, T. J-Aggregates; World Scientific: Singapore, 2012; Volume 2. [Google Scholar]
- Roesch, U.; Yao, S.; Wortmann, R.; Wuerthner, F. Fluorescent H-aggregates of Merocyanine Dyes. Angew. Chem. Int. Ed. 2006, 45, 7026–7030. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Domoto, K.; Isohashi, T.; Kimura, K. In Situ Detection of Birefringent Mesoscopic H and J Aggregates of Thiacarbocyanine Dye in Solution. Langmuir 2005, 21, 1067–1073. [Google Scholar] [CrossRef]
- Oh, J.-W.; Kumazaki, S.; Rubtsov, I.V.; Suzumoto, T.; Tani, T.; Yoshihara, K. Ultrafast Energy Transfer in J-aggregate on AgBr Microcrystals: Its Dependence on Dye Coverage. Chem. Phys. Lett. 2002, 352, 357–362. [Google Scholar] [CrossRef]
- Uppili, S.; Thomas, K.J.; Crompton, E.M.; Ramamurthy, V. Probing Zeolites with Organic Molecules: Supercages of X and Y Zeolites are Superpolar. Langmuir 2000, 16, 265–274. [Google Scholar] [CrossRef]
- Chu, P. The Deammoniation Reaction of Ammonium Y Zeolite. J. Catal. 1976, 43, 346–352. [Google Scholar] [CrossRef]
- Hughes, T.R.; White, H.M. A Study of the Surface Structure of Decationized Y Zeolite by Quantitative Infrared Spectroscopy. J. Phys. Chem. 1967, 71, 2192–2201. [Google Scholar] [CrossRef]
- di Nunzio, M.R.; Caballero-Mancebo, E.; Martín, C.; Cohen, B.; Navarro, M.T.; Corma, A.; Douhal, A. Femto-to Nanosecond Photodynamics of Nile Red in Metal-Ion Exchanged Faujasites. Microporous Mesoporous Mater. 2018, 256, 214–226. [Google Scholar] [CrossRef]
- Organero, J.A.; Tormo, L.; Douhal, A. Caging Ultrafast Proton Transfer and Twisting Motion of 1-Hydroxy-2-Acetonaphthone. Chem. Phys. Lett. 2002, 363, 409–414. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composite | Cavity/Pore diameter (Å) | Total Pore Volume (cm3/g) | DCMloading (1019 DCM molecules/ghost) | Entrapment Efficiency (%) |
|---|---|---|---|---|
| DCM@HY | ~ 11 a | 0.33 b | 6.3 | 91 |
| DCM@NaX | ~ 11 a | 0.29 c | 4.4 | 63 |
| DCM@NaY | ~ 11 a | 0.34 c | 3.9 | 57 |
| DCM@MCM-41 | 21–27 d | 0.98 d | 3.5 | 50 |
| DCM@HY | H-aggregates | J-aggregates | Monomers | Free DCM | |||||
|---|---|---|---|---|---|---|---|---|---|
| [DCM]0/M | λem/nm | τ1 (± 15)/ps | a1/% | τ2 (± 0.07)/ns | a2/% | τ3 (± 0.58)/ns | a3/% | τ4 (± 0.16)/ns | a4/% |
| 10−3 | 435 | 75 | 85 | 0.35 | 14 | 3.83 | 1 | 1.10 | − |
| 450 | 82 | 17 | 1 | − | |||||
| 500 | 62 | 20 | 2 | 16 | |||||
| 550 | 57 | 19 | 3 | 21 | |||||
| 625 | 55 | 15 | 5 | 25 | |||||
| 10−4 | 435 | 79 | 78 | 0.36 | 20 | 3.87 | 2 | 1.10 | − |
| 450 | 75 | 22 | 3 | − | |||||
| 500 | 62 | 26 | 3 | 9 | |||||
| 550 | 57 | 23 | 9 | 11 | |||||
| 625 | 53 | 20 | 10 | 17 | |||||
| 10−5 | 435 | 86 | 73 | 0.37 | 25 | 3.88 | 2 | 1.10 | − |
| 450 | 68 | 28 | 4 | − | |||||
| 500 | 51 | 35 | 8 | 6 | |||||
| 550 | 43 | 34 | 15 | 8 | |||||
| 625 | 34 | 33 | 20 | 13 | |||||
| DCM@host | H-aggregates | J-aggregates | Monomers | ||||
|---|---|---|---|---|---|---|---|
| Host | λem/nm | τ1 (± 15)/ps | a1/% | τ2 (± 0.07)/ns | a2/% | τ3 (± 0.58)/ns | a3/% |
| HY | 435 | 79 | 78 | 0.36 | 20 | 3.87 | 2 |
| 450 | 75 | 22 | 3 | ||||
| NaX | 435 | 96 | 70 | 0.40 | 29 | 2.75 | 1 |
| 450 | 68 | 31 | 1 | ||||
| 475 | 65 | 34 | 1 | ||||
| NaY | 435 | 99 | 65 | 0.36 | 31 | 3.30 | 4 |
| 450 | 59 | 33 | 8 | ||||
| 475 | 55 | 33 | 12 | ||||
| MCM-41 | 450 | 65 | 86 | 0.35 | 13 | 2.46 | 1 |
| 475 | 73 | 24 | 3 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
di Nunzio, M.R.; Perenlei, G.; Douhal, A. Confinement Effect of Micro- and Mesoporous Materials on the Spectroscopy and Dynamics of a Stilbene Derivative Dye. Int. J. Mol. Sci. 2019, 20, 1316. https://doi.org/10.3390/ijms20061316
di Nunzio MR, Perenlei G, Douhal A. Confinement Effect of Micro- and Mesoporous Materials on the Spectroscopy and Dynamics of a Stilbene Derivative Dye. International Journal of Molecular Sciences. 2019; 20(6):1316. https://doi.org/10.3390/ijms20061316
Chicago/Turabian Styledi Nunzio, Maria Rosaria, Ganchimeg Perenlei, and Abderrazzak Douhal. 2019. "Confinement Effect of Micro- and Mesoporous Materials on the Spectroscopy and Dynamics of a Stilbene Derivative Dye" International Journal of Molecular Sciences 20, no. 6: 1316. https://doi.org/10.3390/ijms20061316
APA Styledi Nunzio, M. R., Perenlei, G., & Douhal, A. (2019). Confinement Effect of Micro- and Mesoporous Materials on the Spectroscopy and Dynamics of a Stilbene Derivative Dye. International Journal of Molecular Sciences, 20(6), 1316. https://doi.org/10.3390/ijms20061316