Key Role of Reactive Oxygen Species (ROS) in Indirubin Derivative-Induced Cell Death in Cutaneous T-Cell Lymphoma Cells

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
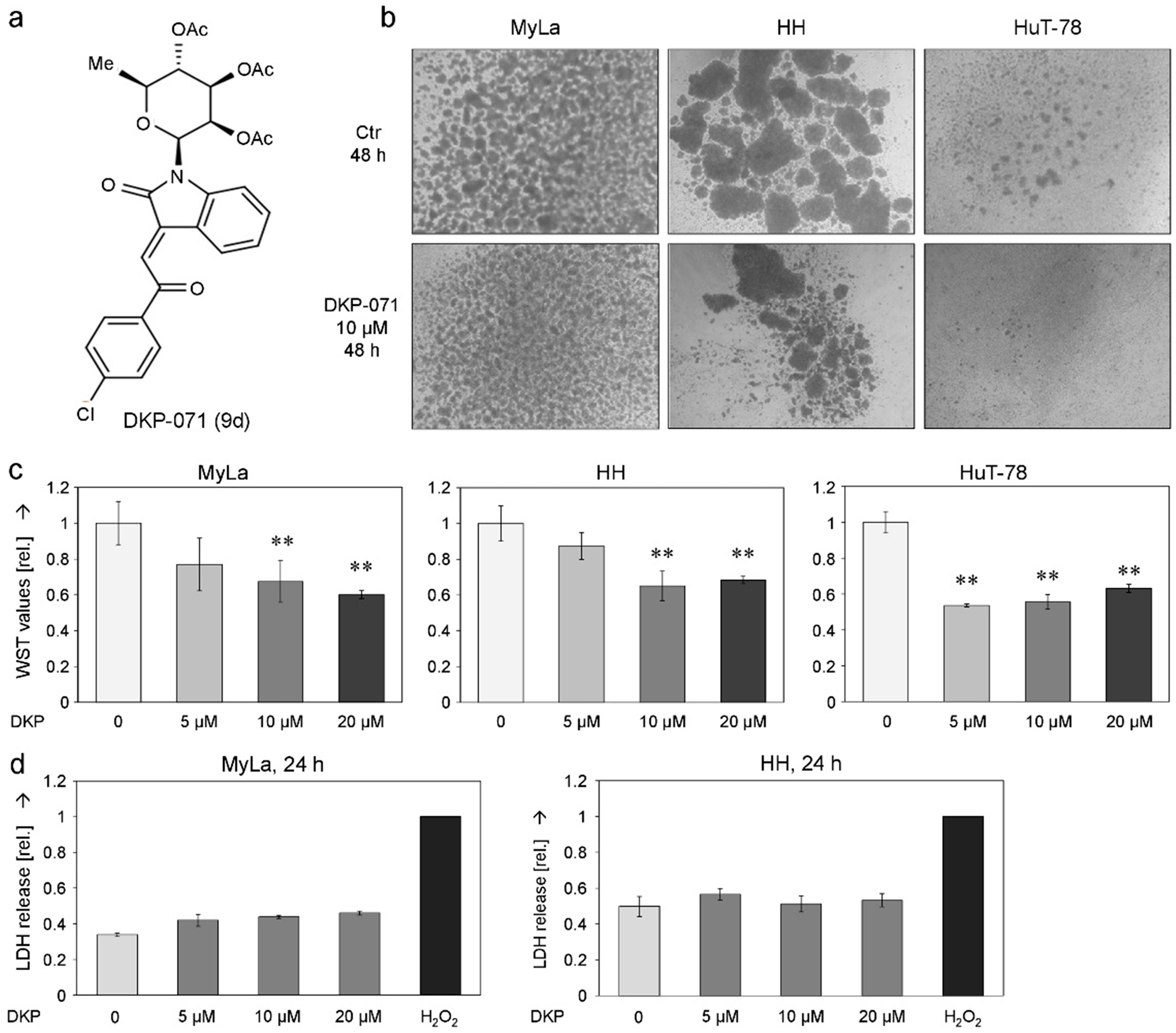
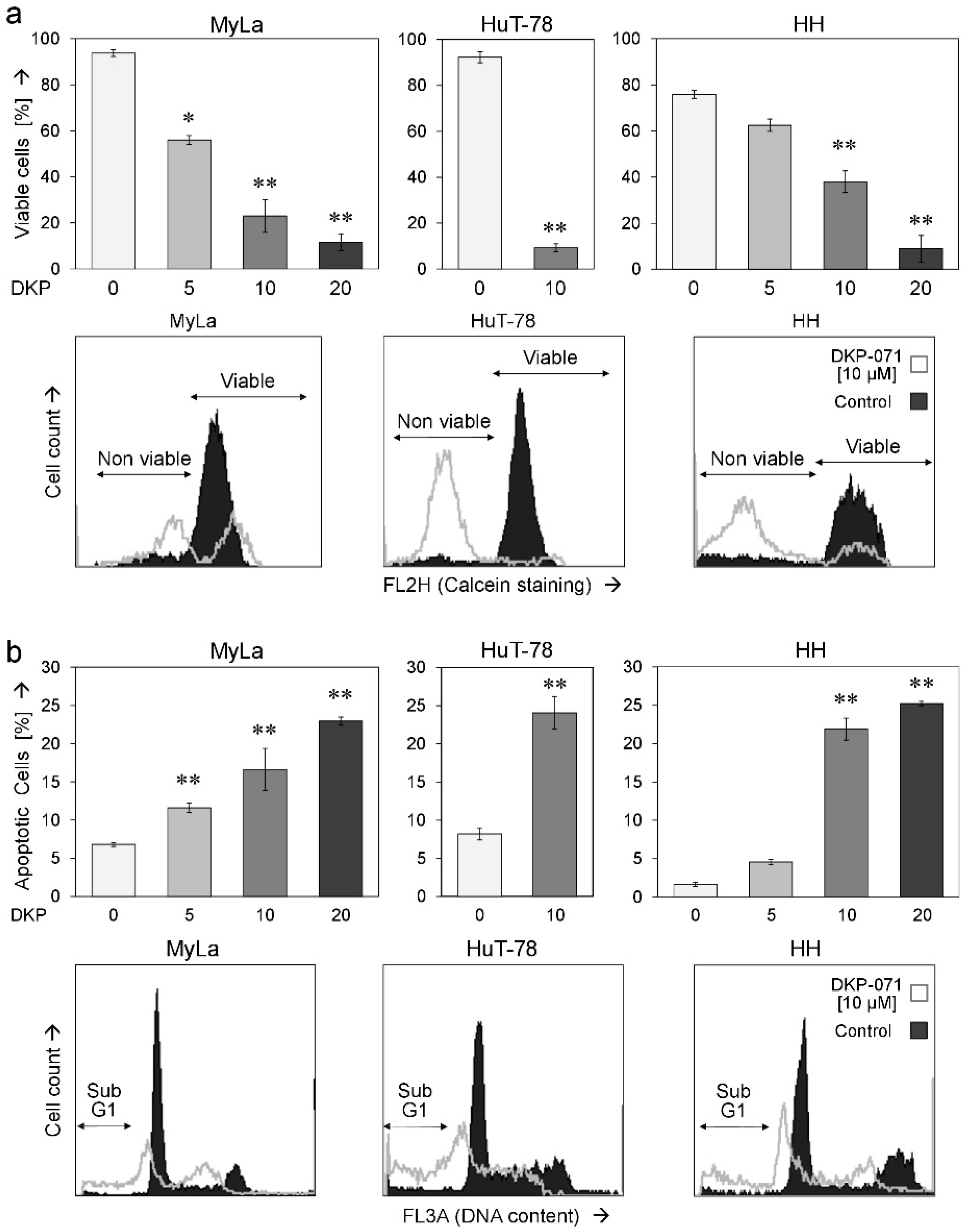
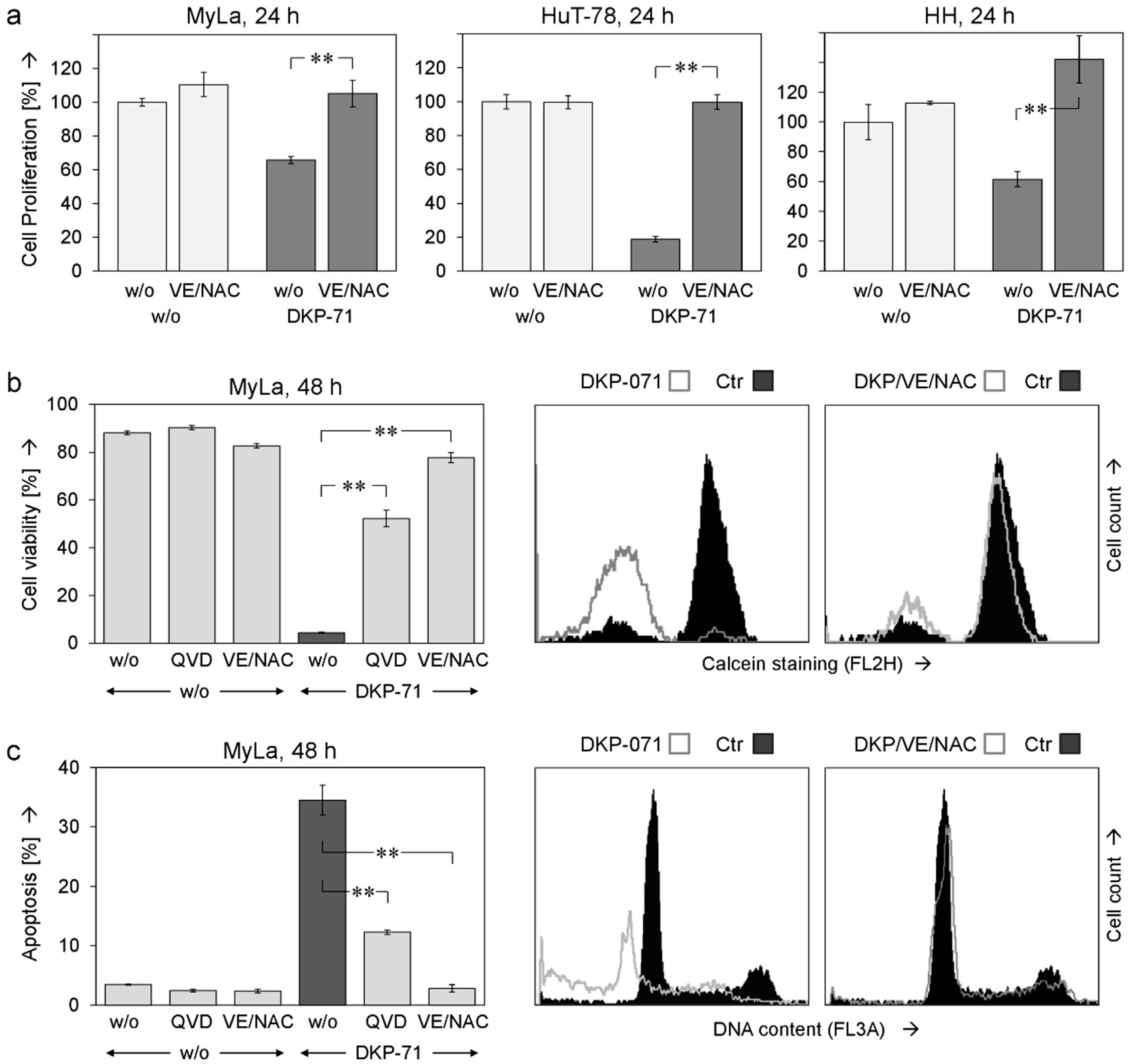
2.1. Decreased Cell Proliferation and Viability Along with Induced Apoptosis by DKP-071
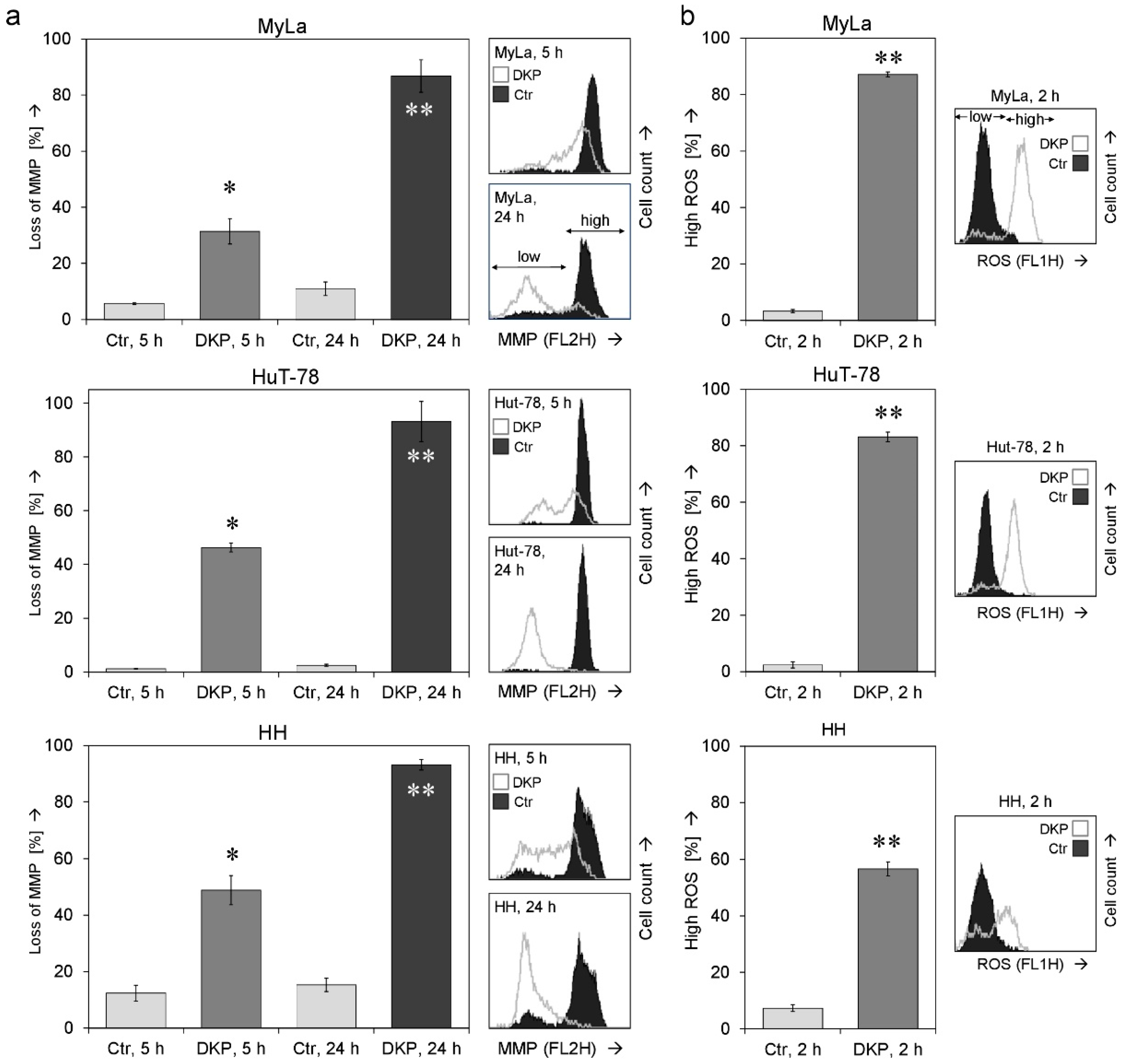
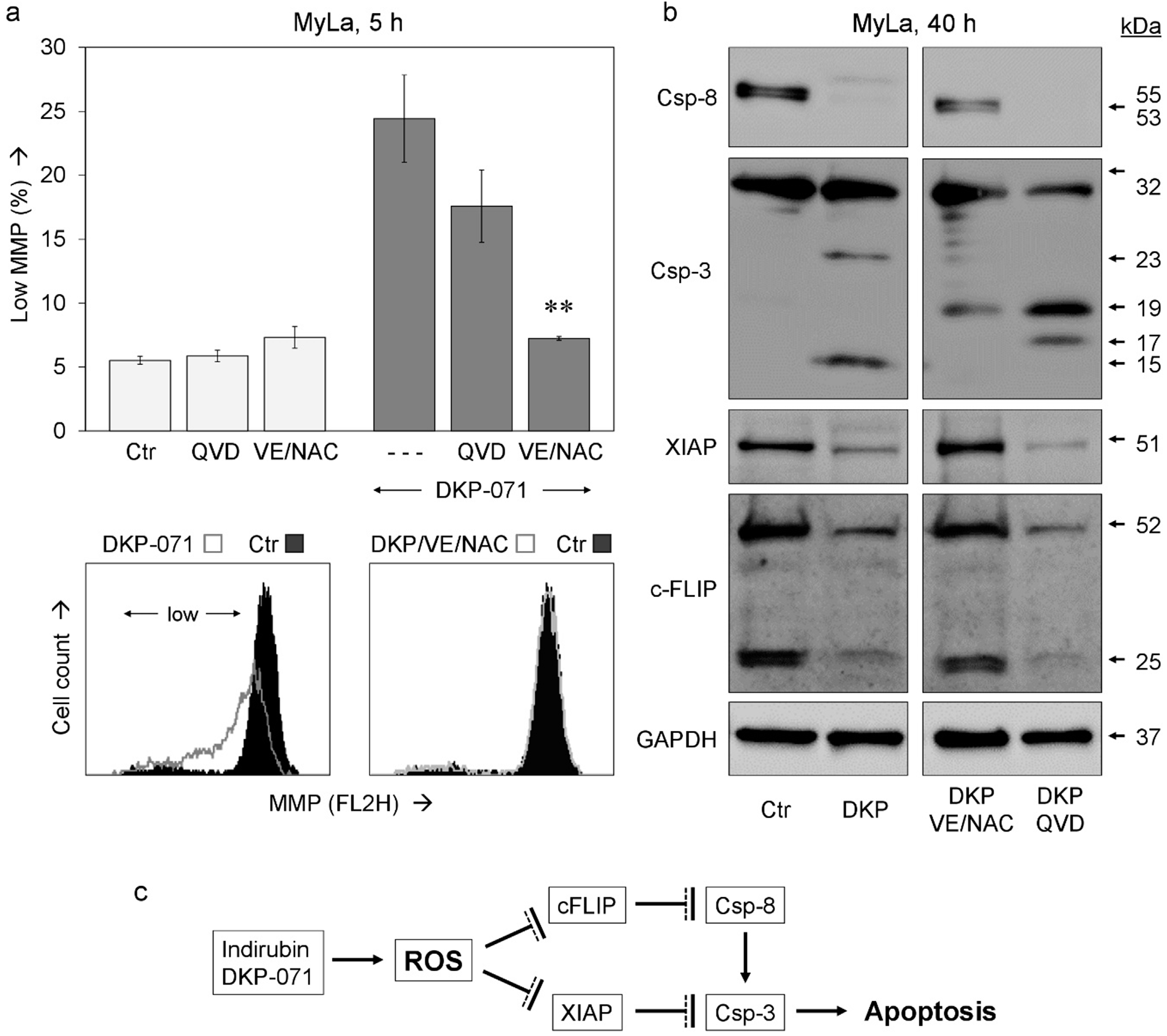
2.2. Changes of Mitochondrial Membrane Potential and ROS Production
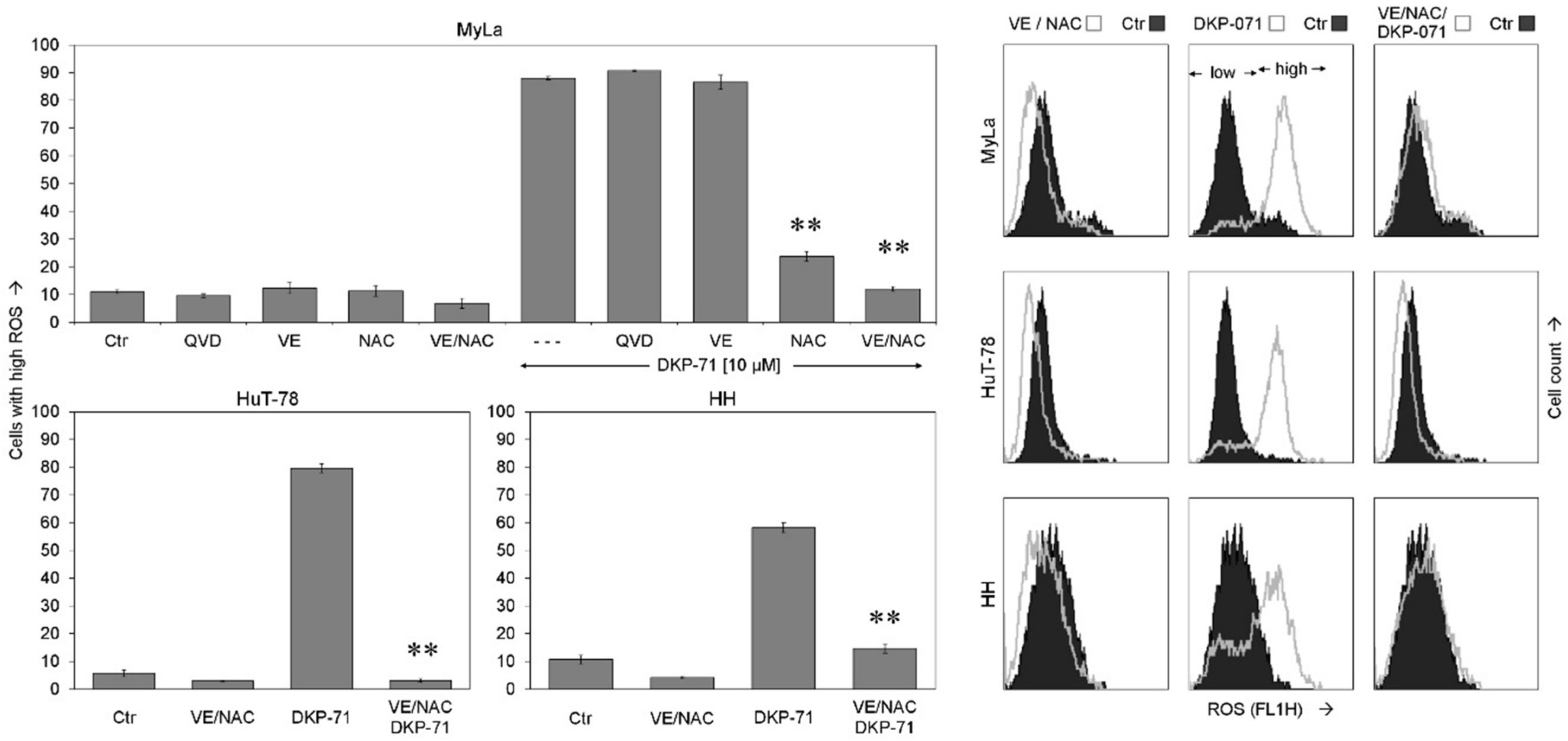
2.3. Critical role of ROS for Proapoptotic Effects of DKP-071
2.4. Role of Caspases and Caspase Antagonistic Proteins
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Assays for Apoptosis, Cytotoxicity, Cell Viability and Cell Proliferation
4.3. Mitochondrial Membrane Potential (MMP) and Reactive Oxygen Species (ROS)
4.4. Western Blotting
4.5. Statistics
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Willemze, R.; Jaffe, E.S.; Burg, G.; Cerroni, L.; Berti, E.; Swerdlow, S.H.; Ralfkiaer, E.; Chimenti, S.; Diaz-Perez, J.L.; Duncan, L.M.; et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005, 105, 3768–3785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jawed, S.I.; Myskowski, P.L.; Horwitz, S.; Moskowitz, A.; Querfeld, C. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome): Part II. Prognosis, management, and future directions. J. Am. Acad. Dermatol. 2014, 70, 223.e1–223.e17. [Google Scholar] [CrossRef] [PubMed]
- Eberle, J.; Kurbanov, B.M.; Hossini, A.M.; Trefzer, U.; Fecker, L.F. Overcoming apoptosis deficiency of melanoma-hope for new therapeutic approaches. Drug Resist. Updates 2007, 10, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Bladon, J.; Taylor, P.C. Extracorporeal photopheresis induces apoptosis in the lymphocytes of cutaneous T-cell lymphoma and graft-versus-host disease patients. Br. J. Haematol. 1999, 107, 707–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krammer, P.H.; Arnold, R.; Lavrik, I.N. Life and death in peripheral T cells. Nat. Rev. Immunol. 2007, 7, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, U.; Janicke, R.U.; Schulze-Osthoff, K. Many cuts to ruin: A comprehensive update of caspase substrates. Cell Death Differ. 2003, 10, 76–100. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Takahashi, R.; Salvesen, G.S.; Reed, J.C. X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997, 388, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Nihal, M.; Siddiqui, J.; Vonderheid, E.C.; Wood, G.S. Low FAS/CD95 expression by CTCL correlates with reduced sensitivity to apoptosis that can be restored by FAS upregulation. J. Investig. Dermatol. 2009, 129, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Braun, F.K.; Fecker, L.F.; Schwarz, C.; Walden, P.; Assaf, C.; Durkop, H.; Sterry, W.; Eberle, J. Blockade of death receptor-mediated pathways early in the signaling cascade coincides with distinct apoptosis resistance in cutaneous T-cell lymphoma cells. J. Investig. Dermatol. 2007, 127, 2425–2437. [Google Scholar] [CrossRef] [PubMed]
- Sors, A.; Jean-Louis, F.; Pellet, C.; Laroche, L.; Dubertret, L.; Courtois, G.; Bachelez, H.; Michel, L. Down-regulating constitutive activation of the NF-kappaB canonical pathway overcomes the resistance of cutaneous T-cell lymphoma to apoptosis. Blood 2006, 107, 2354–2363. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, K.W.; Kaltoft, K.; Mikkelsen, G.; Nielsen, M.; Zhang, Q.; Geisler, C.; Nissen, M.H.; Ropke, C.; Wasik, M.A.; Odum, N. Constitutive STAT3-activation in Sezary syndrome: Tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemic Sezary cells. Leukemia 2001, 15, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Braun, F.K.; Al-Yacoub, N.; Plotz, M.; Mobs, M.; Sterry, W.; Eberle, J. Nonsteroidal anti-inflammatory drugs induce apoptosis in cutaneous T-cell lymphoma cells and enhance their sensitivity for TNF-related apoptosis-inducing ligand. J. Investig. Dermatol. 2012, 132, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Al-Yacoub, N.; Fecker, L.F.; Mobs, M.; Plotz, M.; Braun, F.K.; Sterry, W.; Eberle, J. Apoptosis induction by SAHA in cutaneous T-cell lymphoma cells is related to downregulation of c-FLIP and enhanced TRAIL signaling. J. Investig. Dermatol. 2012, 132, 2263–2274. [Google Scholar] [CrossRef] [PubMed]
- Gahlot, S.; Khan, M.A.; Rishi, L.; Majumdar, S. Pentoxifylline augments TRAIL/Apo2L mediated apoptosis in cutaneous T cell lymphoma (HuT-78 and MyLa) by modulating the expression of antiapoptotic proteins and death receptors. Biochem. Pharmacol. 2010, 80, 1650–1661. [Google Scholar] [CrossRef] [PubMed]
- Quast, S.A.; Berger, A.; Eberle, J. ROS-dependent phosphorylation of Bax by wortmannin sensitizes melanoma cells for TRAIL-induced apoptosis. Cell Death Dis. 2013, 4, e839. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.; Werth, F.; Nguyen, H.A.; Kiecker, F.; Eberle, J. Critical role of reactive oxygen species (ROS) for synergistic enhancement of apoptosis by vemurafenib and the potassium channel inhibitor TRAM-34 in melanoma cells. Cell Death Dis. 2017, 8, e2594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- Salmeron, M.L.; Quintana-Aguiar, J.; De La Rosa, J.V.; Lopez-Blanco, F.; Castrillo, A.; Gallardo, G.; Tabraue, C. Phenalenone-photodynamic therapy induces apoptosis on human tumor cells mediated by caspase-8 and p38-MAPK activation. Mol. Carcinog. 2018, 57, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Salva, K.A.; Kim, Y.H.; Rahbar, Z.; Wood, G.S. Epigenetically Enhanced PDT Induces Significantly Higher Levels of Multiple Extrinsic Pathway Apoptotic Factors than Standard PDT, Resulting in Greater Extrinsic and Overall Apoptosis of Cutaneous T-cell Lymphoma. Photochem. Photobiol. 2018, 94, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Hao, Y.; Liu, B.; Qian, L. Indirubin and meisoindigo in the treatment of chronic myelogenous leukemia in China. Leuk. Lymphoma 2002, 43, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Blazevic, T.; Heiss, E.H.; Atanasov, A.G.; Breuss, J.M.; Dirsch, V.M.; Uhrin, P. Indirubin and Indirubin Derivatives for Counteracting Proliferative Diseases. Evid. Based Complement. Altern. Med. 2015, 2015, 654098. [Google Scholar] [CrossRef] [PubMed]
- Perabo, F.G.; Frossler, C.; Landwehrs, G.; Schmidt, D.H.; von Rucker, A.; Wirger, A.; Muller, S.C. Indirubin-3′-monoxime, a CDK inhibitor induces growth inhibition and apoptosis-independent up-regulation of survivin in transitional cell cancer. Anticancer Res. 2006, 26, 2129–2135. [Google Scholar] [PubMed]
- Meijer, L.; Skaltsounis, A.L.; Magiatis, P.; Polychronopoulos, P.; Knockaert, M.; Leost, M.; Ryan, X.P.; Vonica, C.A.; Brivanlou, A.; Dajani, R.; et al. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 2003, 10, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Sorensen, V.; Jin, Y.; Suo, Z.; Wiedlocha, A. Indirubin-3′-monoxime inhibits autophosphorylation of FGFR1 and stimulates ERK1/2 activity via p38 MAPK. Oncogene 2007, 26, 6372–6385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Du, Z.; Zhuang, Z.; Wang, Y.; Wang, F.; Liu, S.; Wang, H.; Feng, H.; Li, H.; Wang, L.; et al. E804 induces growth arrest, differentiation and apoptosis of glioblastoma cells by blocking Stat3 signaling. J. Neurooncol. 2015, 125, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Quast, S.A.; Plotz, M.; Hein, M.; Kunz, M.; Langer, P.; Eberle, J. Sensitization of melanoma cells for death ligand-induced apoptosis by an indirubin derivative—Enhancement of both extrinsic and intrinsic apoptosis pathways. Biochem. Pharmacol. 2011, 81, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhivkova, V.; Kiecker, F.; Langer, P.; Eberle, J. Crucial role of reactive oxygen species (ROS) for the proapoptotic effects of indirubin derivative DKP-073 in melanoma cells. Mol. Carcinog. 2019, 58, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Libnow, S.; Methling, K.; Hein, M.; Michalik, D.; Harms, M.; Wende, K.; Flemming, A.; Kockerling, M.; Reinke, H.; Bednarski, P.J.; et al. Synthesis of indirubin-N′-glycosides and their anti-proliferative activity against human cancer cell lines. Bioorg. Med. Chem. 2008, 16, 5570–5583. [Google Scholar] [CrossRef] [PubMed]
- Kunz, M.; Driller, K.M.; Hein, M.; Libnow, S.; Hohensee, I.; Ramer, R.; Hinz, B.; Berger, A.; Eberle, J.; Langer, P. Synthesis of thia-analogous indirubin N-Glycosides and their influence on melanoma cell growth and apoptosis. ChemMedChem 2010, 5, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Kleeblatt, D.; Becker, M.; Plötz, M.; Schonherr, M.; Villinger, A.; Hein, M.; Eberle, J.; Kunz, M.; Rahman, Q.; Langer, P. Synthesis and Bioactivity of N-glycosylated 3-(2-Oxo-2- arylethylidene)-indolin-2-ones. RSC Adv. 2015, 5, 20623–20633. [Google Scholar] [CrossRef]
- Bang, K.; Lund, M.; Mogensen, S.C.; Thestrup-Pedersen, K. In vitro culture of skin-homing T lymphocytes from inflammatory skin diseases. Exp. Dermatol. 2005, 14, 391–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drexler, H.G.; Pommerenke, C.; Eberth, S.; Nagel, S. Hodgkin lymphoma cell lines: To separate the wheat from the chaff. Biol. Chem. 2018, 399, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Braun, F.K.; Hirsch, B.; Al-Yacoub, N.; Durkop, H.; Assaf, C.; Kadin, M.E.; Sterry, W.; Eberle, J. Resistance of cutaneous anaplastic large-cell lymphoma cells to apoptosis by death ligands is enhanced by CD30-mediated overexpression of c-FLIP. J. Investig. Dermatol. 2010, 130, 826–840. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Zhang, Y.; Jie, X.M.; She, J.; Dongye, G.Z.; Zhong, Y.; Deng, Y.Y.; Wang, J.; Guo, B.Y.; Chen, L.M. Ruthenium(II) salicylate complexes inducing ROS-mediated apoptosis by targeting thioredoxin reductase. J. Inorg. Biochem. 2019, 193, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, T.; Hagihara, K.; Satoh, R.; Sugiura, R. More than Just an Immunosuppressant: The Emerging Role of FTY720 as a Novel Inducer of ROS and Apoptosis. Oxid. Med. Cell. Longev. 2018, 2018, 4397159. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Washington, A.; Leaf, R.K.; Bhargava, P.; Clark, R.A.; Kupper, T.S.; Stroopinsky, D.; Pyzer, A.; Cole, L.; Nahas, M.; et al. Decitabine Priming Enhances Mucin 1 Inhibition Mediated Disruption of Redox Homeostasis in Cutaneous T-Cell Lymphoma. Mol. Cancer Ther. 2017, 16, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Sun, J.; Qiu, L.; Fu, W.; Wang, A.; Liu, X.; Yang, Y.; Kadin, M.E.; Tu, P.; Wang, Y. Dual Role of EZH2 in Cutaneous Anaplastic Large Cell Lymphoma: Promoting Tumor Cell Survival and Regulating Tumor Microenvironment. J. Investig. Dermatol. 2017, 138, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Alexander-Savino, C.V.; Hayden, M.S.; Richardson, C.; Zhao, J.; Poligone, B. Doxycycline is an NF-kappaB inhibitor that induces apoptotic cell death in malignant T-cells. Oncotarget 2016, 7, 75954–75967. [Google Scholar] [CrossRef] [PubMed]
- Kaltoft, K.; Bisballe, S.; Dyrberg, T.; Boel, E.; Rasmussen, P.B.; Thestrup-Pedersen, K. Establishment of two continuous T-cell strains from a single plaque of a patient with mycosis fungoides. In Vitro Cell. Dev. Biol. 1992, 28A, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.E.; Ruscetti, F.W.; Mier, J.W.; Gazdar, A.; Gallo, R.C. Human cutaneous T cell lymphoma and leukemia cell lines produce and respond to T cell growth factor. J. Exp. Med. 1981, 154, 1403–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starkebaum, G.; Loughran, T.P., Jr.; Waters, C.A.; Ruscetti, F.W. Establishment of an IL-2 independent, human T-cell line possessing only the p70 IL-2 receptor. Int. J. Cancer 1991, 49, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef] [PubMed]
- Creed, S.; McKenzie, M. Measurement of Mitochondrial Membrane Potential with the Fluorescent Dye Tetramethylrhodamine Methyl Ester (TMRM). Methods Mol. Biol. 2019, 1928, 69–76. [Google Scholar] [PubMed]
- Eberle, J.; Fecker, L.F.; Hossini, A.M.; Wieder, T.; Daniel, P.T.; Orfanos, C.E.; Geilen, C.C. CD95/Fas signaling in human melanoma cells: Conditional expression of CD95L/FasL overcomes the intrinsic apoptosis resistance of malignant melanoma and inhibits growth and progression of human melanoma xenotransplants. Oncogene 2003, 22, 9131–9141. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soltan, M.Y.; Sumarni, U.; Assaf, C.; Langer, P.; Reidel, U.; Eberle, J. Key Role of Reactive Oxygen Species (ROS) in Indirubin Derivative-Induced Cell Death in Cutaneous T-Cell Lymphoma Cells. Int. J. Mol. Sci. 2019, 20, 1158. https://doi.org/10.3390/ijms20051158
Soltan MY, Sumarni U, Assaf C, Langer P, Reidel U, Eberle J. Key Role of Reactive Oxygen Species (ROS) in Indirubin Derivative-Induced Cell Death in Cutaneous T-Cell Lymphoma Cells. International Journal of Molecular Sciences. 2019; 20(5):1158. https://doi.org/10.3390/ijms20051158
Chicago/Turabian StyleSoltan, Marwa Y., Uly Sumarni, Chalid Assaf, Peter Langer, Ulrich Reidel, and Jürgen Eberle. 2019. "Key Role of Reactive Oxygen Species (ROS) in Indirubin Derivative-Induced Cell Death in Cutaneous T-Cell Lymphoma Cells" International Journal of Molecular Sciences 20, no. 5: 1158. https://doi.org/10.3390/ijms20051158
APA StyleSoltan, M. Y., Sumarni, U., Assaf, C., Langer, P., Reidel, U., & Eberle, J. (2019). Key Role of Reactive Oxygen Species (ROS) in Indirubin Derivative-Induced Cell Death in Cutaneous T-Cell Lymphoma Cells. International Journal of Molecular Sciences, 20(5), 1158. https://doi.org/10.3390/ijms20051158