VKORC1 and CYP2C9 Polymorphisms: A Case Report in a Dutch Family with Pulmonary Fibrosis


 ,
,
Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Antoniou, K.M.; Margaritopoulos, G.A.; Tomassetti, S.; Bonella, F.; Costabel, U.; Poletti, V. Interstitial lung disease. Eur. Respir. Rev. 2014, 23, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, W.A.; Cavazza, A.; Rossi, G.; Bonella, F.; Sverzellati, N.; Spagnolo, P. Differential diagnosis of usual interstitial pneumonia: When is it truly idiopathic? Eur. Respir. Rev. 2014, 23, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Sverzellati, N.; Rossi, G. IPF in 2016: Towards a better diagnosis. Lancet. Respir. Med. 2016, 4, 945–947. [Google Scholar] [CrossRef]
- Bonham, C.A.; Strek, M.E.; Patterson, K.C. From granuloma to fibrosis: Sarcoidosis associated pulmonary fibrosis. Curr. Opin. Pulm. Med. 2016, 22, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.P.; Speer, M.C.; Loyd, J.E.; Brown, K.K.; Herron, A.; Slifer, S.H.; Burch, L.H.; Wahidi, M.M.; Phillips, J.A., 3rd; Sporn, T.A.; et al. Clinical and pathologic features of familial interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2005, 172, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sancho, C.; Buendia-Roldan, I.; Fernandez-Plata, M.R.; Navarro, C.; Perez-Padilla, R.; Vargas, M.H.; Loyd, J.E.; Selman, M. Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respir. Med. 2011, 105, 1902–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, S.; Tanaka, T.; Ishida, M.; Kinoshita, A.; Fukuoka, J.; Takaki, M.; Sakamoto, N.; Ishimatsu, Y.; Kohno, S.; Hayashi, T.; et al. Surfactant protein C G100S mutation causes familial pulmonary fibrosis in Japanese kindred. Eur. Respir. J. 2011, 38, 861–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, T.W.; van Moorsel, C.H.M.; Borie, R.; Crestani, B. Pulmonary phenotypes associated with genetic variation in telomere-related genes. Curr. Opin. Pulm. Med. 2018, 24, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Borie, R.; Kannengiesser, C.; Nathan, N.; Tabeze, L.; Pradere, P.; Crestani, B. Familial pulmonary fibrosis. Rev. Mal. Respir. 2015, 32, 413–434. [Google Scholar] [CrossRef] [PubMed]
- Borie, R.; Tabeze, L.; Thabut, G.; Nunes, H.; Cottin, V.; Marchand-Adam, S.; Prevot, G.; Tazi, A.; Cadranel, J.; Mal, H.; et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur. Respir. J. 2016, 48, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, W.A.; Agostini, C.; Antoniou, K.M.; Bouros, D.; Chambers, R.C.; Cottin, V.; Egan, J.J.; Lambrecht, B.N.; Lories, R.; Parfrey, H.; et al. The pathogenesis of pulmonary fibrosis: A moving target. Eur. Respir. J. 2013, 41, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.; Mazzei, M.A.; Squitieri, N.C.; Bargagli, E.; Refini, R.M.; Fossi, A.; Volterrani, L.; Rottoli, P. Familial pulmonary fibrosis: Clinical and radiological characteristics and progression analysis in different high resolution-CT patterns. Respir.Med. 2017, 126, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.A.; Sverzellati, N.; Travis, W.D.; Brown, K.K.; Colby, T.V.; Galvin, J.R.; Goldin, J.G.; Hansell, D.M.; Inoue, Y.; Johkoh, T.; et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet. Respir. Med. 2018, 6, 138–153. [Google Scholar] [CrossRef]
- Lawson, W.E.; Loyd, J.E.; Degryse, A.L. Genetics in pulmonary fibrosis—familial cases provide clues to the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Med. Sci. 2011, 341, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Van Moorsel, C.H.; van Oosterhout, M.F.; Barlo, N.P.; de Jong, P.A.; van der Vis, J.J.; Ruven, H.J.; van Es, H.W.; van den Bosch, J.M.; Grutters, J.C. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am. J. Respir. Crit. Care Med. 2010, 182, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Ravaglia, C.; Tomassetti, S.; Gurioli, C.; Piciucchi, S.; Dubini, A.; Casoni, G.L.; Romagnoli, M.; Carloni, A.; Tantalocco, P.; Buccioli, M.; et al. Features and outcome of familial idiopathic pulmonary fibrosis. Sarcoidosis. Vasc. Diffuse Lung Dis. 2014, 31, 28–36. [Google Scholar] [PubMed]
- Spagnolo, P.; Grunewald, J.; du Bois, R.M. Genetic determinants of pulmonary fibrosis: Evolving concepts. Lancet. Respir. Med. 2014, 2, 416–428. [Google Scholar] [CrossRef]
- Van Moorsel, C.H.M. Trade-offs in aging lung diseases: A review on shared but opposite genetic risk variants in idiopathic pulmonary fibrosis, lung cancer and chronic obstructive pulmonary disease. Curr. Opin. Pulm. Med. 2018, 24, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Downey, G.P. Resolving the scar of pulmonary fibrosis. N. Engl. J. Med. 2011, 365, 1140–1141. [Google Scholar] [CrossRef] [PubMed]
- Wijnen, P.A.; Linssen, C.F.; Haenen, G.R.; Bekers, O.; Drent, M. Variant VKORC1 and CYP2C9 alleles in patients with diffuse alveolar hemorrhage caused by oral anticoagulants. Mol. Diagn. Ther. 2010, 14, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, H.; Girdhar, A.; Usman, F.; Cury, J.; Bajwa, A. Approach to acute exacerbation of idiopathic pulmonary fibrosis. Ann. Thorac. Med. 2013, 8, 71–77. [Google Scholar] [PubMed]
- Drent, M.; Wessels, S.; Jacobs, J.A.; Thijssen, H. Association of diffuse alveolar haemorrhage with acquired vitamin K deficiency. Respiration 2000, 67, 697. [Google Scholar] [CrossRef] [PubMed]
- Wijnen, P.A.; Verschakelen, J.A.; Bast, A.; Bekers, O.; Drent, M. Diffuse alveolar hemorrhage in coumarin users: A fibrosing interstitial pneumonia trigger? Lung 2013, 191, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Worldwide Distribution of Cytochrome P450 Alleles: A Meta-analysis of Population-scale Sequencing Projects. Clin. Pharmacol. Ther. 2017, 102, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.Y.; Chen, J.J.; Lee, M.T.; Wung, J.C.; Chen, Y.F.; Charng, M.J.; Lu, M.J.; Hung, C.R.; Wei, C.Y.; Chen, C.H.; et al. A novel functional VKORC1 promoter polymorphism is associated with inter-individual and inter-ethnic differences in warfarin sensitivity. Hum. Mol. Genet. 2005, 14, 1745–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drent, M.; Wijnen, P.; Bast, A. Pharmacogenetic variants and vitamin K deficiency: A risk factor or trigger for fibrosing interstitial pneumonias? Curr. Opin. Pulm. Med. 2018, 24, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Choi, A.M. Cytoprotection of heme oxygenase-1/carbon monoxide in lung injury. Proc. Am. Thor. Soc. 2005, 2, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Bast, A.; Weseler, A.R.; Haenen, G.R.; den Hartog, G.J. Oxidative stress and antioxidants in interstitial lung disease. Curr. Opin. Pulm. Med. 2010, 16, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.A.; Khasawneh, R.; Peter, I.; Kornreich, R.; Desnick, R.J. Combined CYP2C9, VKORC1 and CYP4F2 frequencies among racial and ethnic groups. Pharmacogenomics 2010, 11, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.; Scott, S.D.; Mackie, I.J.; Shearer, M.; Bax, R.; Karran, S.J.; Machin, S.J. The development of hypoprothrombinaemia following antibiotic therapy in malnourished patients with low serum vitamin K1 levels. Br. J. Haematol. 1988, 68, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, H.; Hatta, K.; Usui, C.; Ito, M.; Kita, Y.; Arai, H. Vitamin K deficiency due to prolongation of antibiotic treatment and decrease in food intake in a catatonia patient. Psychosomatics 2011, 52, 486–487. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gray, J.P.; Mishin, V.; Heck, D.E.; Laskin, D.L.; Laskin, J.D. Role of cytochrome P450 reductase in nitrofurantoin-induced redox cycling and cytotoxicity. Free Radic. Biol. Med. 2008, 44, 1169–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, S.; den Hartog, G.J.; Bast, A. Superoxide radicals increase transforming growth factor-β1 and collagen release from human lung fibroblasts via cellular influx through chloride channels. Toxicol. Appl. Pharmacol. 2009, 237, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Nishiumi, S.; Nishida, M.; Mizushina, Y.; Kobayashi, K.; Masuda, A.; Fujita, T.; Morita, Y.; Mizuno, S.; Kutsumi, H.; et al. Vitamin K3 attenuates lipopolysaccharide-induced acute lung injury through inhibition of nuclear factor-κB activation. Clin. Exp. Immunol. 2010, 160, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Drent, M.; Wijnen, P.; Bast, A. Interstitial lung damage due to cocaine abuse: Pathogenesis, pharmacogenomics and therapy. Curr. Med. Chem. 2012, 19, 5607–5611. [Google Scholar] [CrossRef] [PubMed]
- Hoult, J.R.; Paya, M. Pharmacological and biochemical actions of simple coumarins: Natural products with therapeutic potential. Gen. Pharmacol. 1996, 27, 713–722. [Google Scholar] [CrossRef]
- Wijnen, P.A.; Bekers, O.; Drent, M. Development of cocaine-induced interstitial lung damage in two CYP2C and VKORC1 variant allele carriers. Mol. Diagn. Ther. 2011, 15, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Cousin, M.A.; Matey, E.T.; Blackburn, P.R.; Boczek, N.J.; McAllister, T.M.; Kruisselbrink, T.M.; Babovic-Vuksanovic, D.; Lazaridis, K.N.; Klee, E.W. Pharmacogenomic findings from clinical whole exome sequencing of diagnostic odyssey patients. Mol. Genet. Genomic. Med. 2017, 5, 269–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laine, K.; Yasar, U.; Widen, J.; Tybring, G. A screening study on the liability of eight different female sex steroids to inhibit CYP2C9, 2C19 and 3A4 activities in human liver microsomes. Pharmacol. Toxicol. 2003, 93, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Villablanca, A.C.; Jayachandran, M.; Banka, C. Atherosclerosis and sex hormones: Current concepts. Clin. Sci. 2010, 119, 493–513. [Google Scholar] [CrossRef] [PubMed]
- Varone, F.; Montemurro, G.; Macagno, F.; Calvello, M.; Conte, E.; Intini, E.; Iovene, B.; Leone, P.M.; Mari, P.V.; Richeldi, L. Investigational drugs for idiopathic pulmonary fibrosis. Expert. Opin. Investig. Drugs 2017, 26, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Noth, I.; Anstrom, K.J.; Calvert, S.B.; de Andrade, J.; Flaherty, K.R.; Glazer, C.; Kaner, R.J.; Olman, M.A. A Placebo-Controlled Randomized Trial of Warfarin in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 88–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuter, M.; Wijsenbeek, M.S.; Vasakova, M.; Spagnolo, P.; Kolb, M.; Costabel, U.; Weycker, D.; Kirchgaessler, K.U.; Maher, T.M. Unfavourable effects of medically indicated oral anticoagulants on survival in idiopathic pulmonary fibrosis. Eur. Respir. J. 2016, 47, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Piscaer, I.; Wouters, E.F.M.; Vermeer, C.; Janssens, W.; Franssen, F.M.E.; Janssen, R. Vitamin K deficiency: The linking pin between COPD and cardiovascular diseases? Respir. Res. 2017, 18, 189. [Google Scholar] [CrossRef] [PubMed]
- Crowther, M.A.; Garcia, D.; Ageno, W.; Wang, L.; Witt, D.M.; Clark, N.P.; Blostein, M.D.; Kahn, S.R.; Schulman, S.; Kovacs, M.; et al. Oral vitamin K effectively treats international normalised ratio (INR) values in excess of 10. Results of a prospective cohort study. Thromb. Haemost. 2010, 104, 118–121. [Google Scholar] [PubMed]
- Sproll, C.; Ruge, W.; Andlauer, C.; Godelmann, R.; Lachenmeier, D.W. HPLC analysis and safety assessment of coumarin in foods. Food Chem. 2008, 109, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Kruger, S.; Winheim, L.; Morlock, G.E. Planar chromatographic screening and quantification of coumarin in food, confirmed by mass spectrometry. Food Chem. 2018, 239, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; He, X.; Zhong, P.; Zhao, J.; Huang, C.; Hu, Z. A Metabolism-Based Synergy for Total Coumarin Extract of Radix Angelicae Dahuricae and Ligustrazine on Migraine Treatment in Rats. Molecules 2018, 23, 5. [Google Scholar] [CrossRef] [PubMed]
- Mooiman, K.D.; Goey, A.K.; Huijbregts, T.J.; Maas-Bakker, R.F.; Beijnen, J.H.; Schellens, J.H.; Meijerman, I. The in-vitro effect of complementary and alternative medicines on cytochrome P450 2C9 activity. J. Pharm. Pharmacol. 2014, 66, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Borlak, J.; Thum, T. Identification of major CYP2C9 and CYP2C19 polymorphisms by fluorescence resonance energy transfer analysis. Clin. Chem. 2002, 48, 1592–1594. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
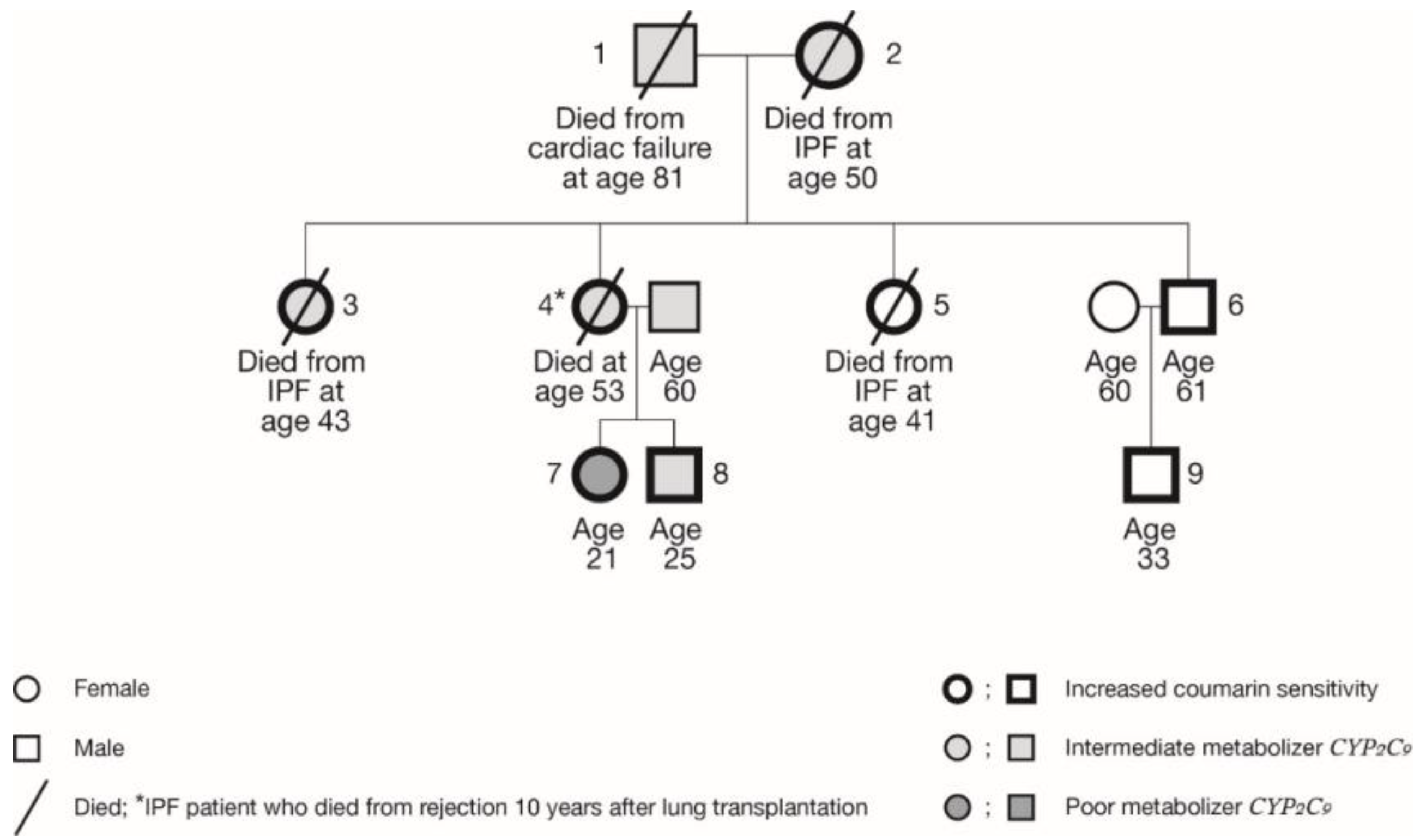
| Family Member | Year of Birth | Age (yr) at Diagnosis of IPF | Age (yr) Deceased | Current Age (yr) | Sex | CYP2C9 | MetabolicfunctionCyp2c9 Enzyme | VKORC1 | Coumarin Sensitivity VKORC1 Enzyme | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1934 | NA | 81 | NA | m | *1/*2 | IM | CC | GG | normal |
| 2 | 1933 | 39 | 50 | NA | f | *1/*3 | IM | TT | AA | high |
| 3 | 1959 | 38 | 43 | NA | f | *1/*3 | IM | CT | GA | increased |
| 4 | 1960 | 25 | 53 | NA | f | *1/*2 | IM | CT | GA | increased |
| 5 | 1964 | 41 | 45 | NA | f | *1/*1 | EM | CT | GA | increased |
| 6 | 1957 | NA | NA | 61 | m | *1/*1 | EM | CT | GA | increased |
| 7 | 1997 | NA | NA | 21 | f | *2/*2 | PM | CT | GA | increased |
| 8 | 1993 | NA | NA | 25 | m | *1/*2 | IM | CT | GA | increased |
| 9 | 1985 | NA | NA | 33 | m | *1/*1 | EM | CT | GA | increased |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wijnen, P.; Drent, M.; Bekers, O.; Verschakelen, J.; Bast, A. VKORC1 and CYP2C9 Polymorphisms: A Case Report in a Dutch Family with Pulmonary Fibrosis. Int. J. Mol. Sci. 2019, 20, 1160. https://doi.org/10.3390/ijms20051160
Wijnen P, Drent M, Bekers O, Verschakelen J, Bast A. VKORC1 and CYP2C9 Polymorphisms: A Case Report in a Dutch Family with Pulmonary Fibrosis. International Journal of Molecular Sciences. 2019; 20(5):1160. https://doi.org/10.3390/ijms20051160
Chicago/Turabian StyleWijnen, Petal, Marjolein Drent, Otto Bekers, Johny Verschakelen, and Aalt Bast. 2019. "VKORC1 and CYP2C9 Polymorphisms: A Case Report in a Dutch Family with Pulmonary Fibrosis" International Journal of Molecular Sciences 20, no. 5: 1160. https://doi.org/10.3390/ijms20051160
APA StyleWijnen, P., Drent, M., Bekers, O., Verschakelen, J., & Bast, A. (2019). VKORC1 and CYP2C9 Polymorphisms: A Case Report in a Dutch Family with Pulmonary Fibrosis. International Journal of Molecular Sciences, 20(5), 1160. https://doi.org/10.3390/ijms20051160