The Contribution of Genetic Factors to Cognitive Impairment and Dementia: Apolipoprotein E Gene, Gene Interactions, and Polygenic Risk

 ,
,
Abstract
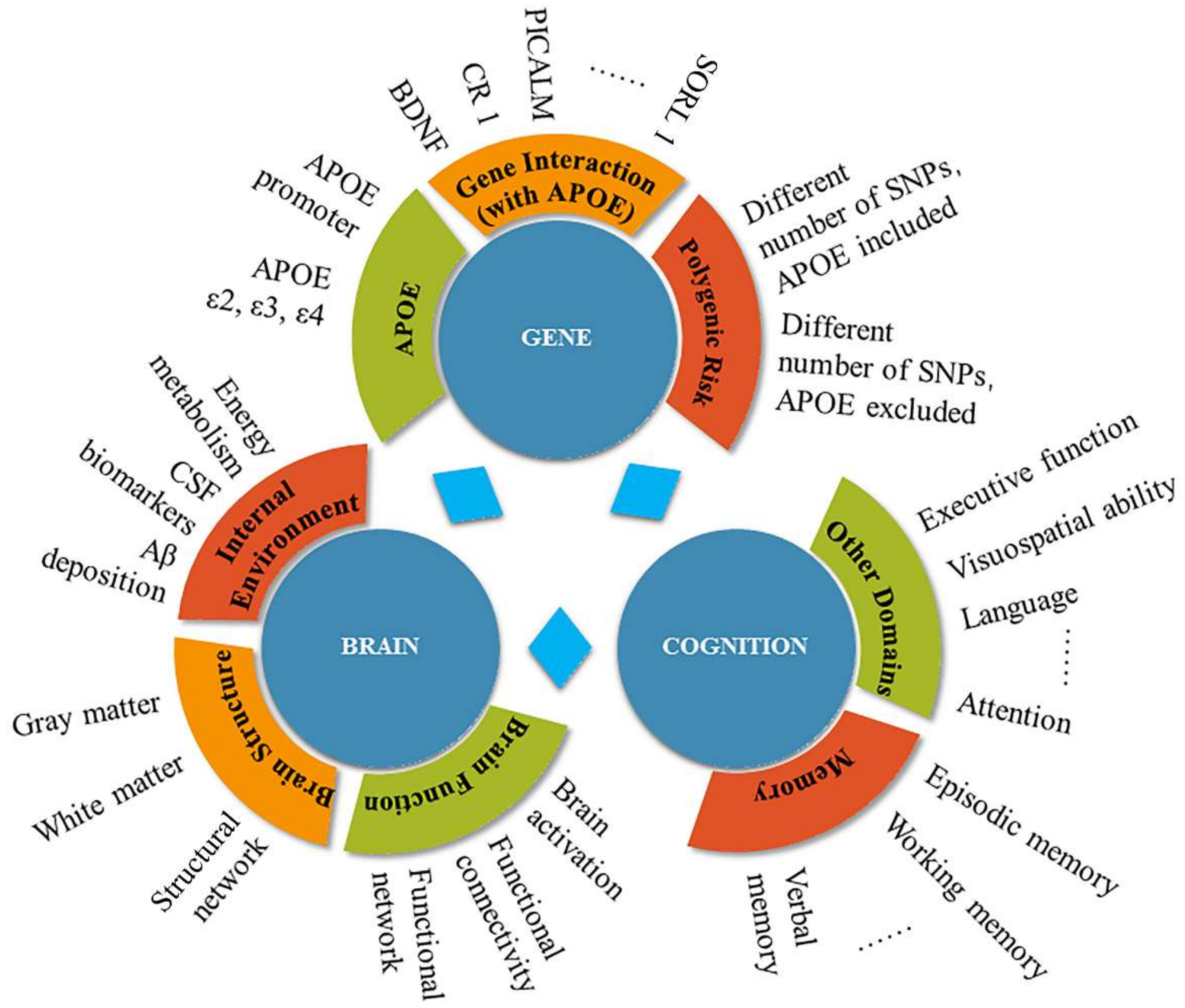
:1. Introduction
1.1. Alzheimer’s Disease and Genetics
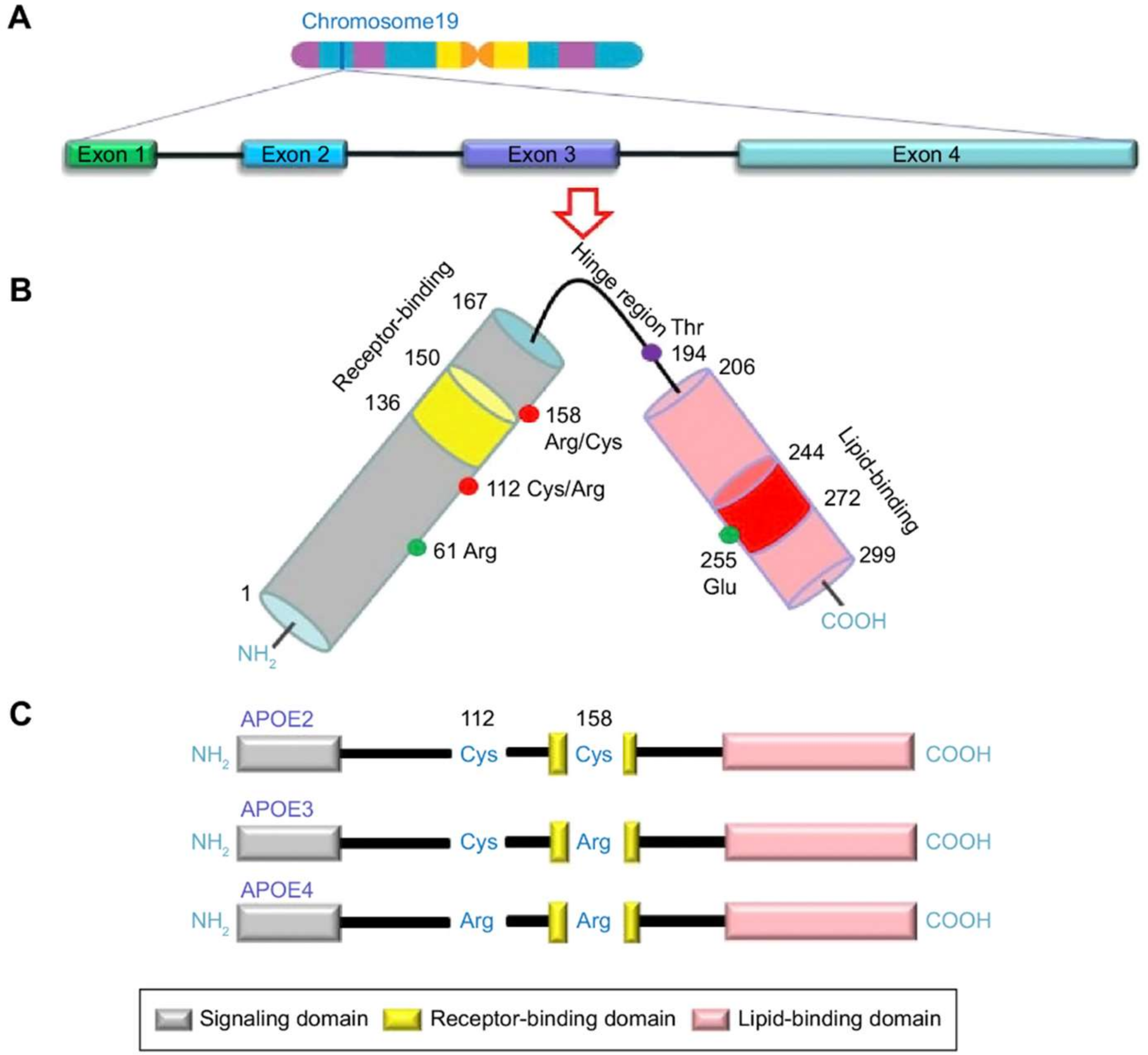
1.2. APOE: Risk Factor for AD
2. The Effects of the APOE Gene on Cognitive Function and Dementia
2.1. APOE ε4 Allele
2.2. Promoter Polymorphisms of the APOE Gene
2.3. Genetic Association with the APOE Gene
2.4. Polygenic Risk Factors for Cognitive Decline
3. The Effects of the APOE Gene on Brain Function
3.1. APOE ε4 Allele
3.2. Promoter Polymorphisms of the APOE Gene
3.3. Genetic Association with the APOE Gene
3.4. Polygenic Risk for the Brain
4. Discussion
4.1. Study Sample
4.1.1. Age
4.1.2. Family History
4.1.3. Other Diseases
4.2. Methodological Issue
4.2.1. Uniformity
4.2.2. Study Design
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Patterson, C. World Alzheimer Report 2018—The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International (ADI): London, UK, 2018. [Google Scholar]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Campion, D.; Dumanchin, C.; Hannequin, D.; Dubois, B.; Belliard, S.; Puel, M.; Thomas-Anterion, C.; Michon, A.; Martin, C.; Charbonnier, F. Early-Onset Autosomal Dominant Alzheimer Disease: Prevalence, Genetic Heterogeneity, and Mutation Spectrum. Am. J. Hum. Genet. 1999, 65, 664–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Kamboh, M.I. Molecular genetics of late-onset Alzheimer’s disease. Ann. Hum. Genet. 2012, 68, 381–404. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C. Meta-analysis in more than 74,000 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Alzheimers Dement. 2013, 9, 1452–1458. [Google Scholar] [CrossRef]
- Avila, J.; Gomez-Ramos, A.; Bolos, M. AD genetic risk factors and tau spreading. Front. Aging Neurosci. 2015, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Goate, A.M. Alzheimer’s Disease Risk Genes and Mechanisms of Disease Pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zannis, V.I.; Just, P.W.; Breslow, J.L. Human apolipoprotein E isoprotein subclasses are genetically determined. Am. J. Hum. Genet. 1981, 33, 11. [Google Scholar] [PubMed]
- Singh, P.P.; Singh, M.; Mastana, S.S. APOE distribution in world populations with new data from India and the UK. Ann. Hum. Biol. 2006, 33, 279–308. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, L.U.; Gerdes, C.; Hansen, P.S.; Klausen, I.C.; Faergeman, O.; Dyerberg, J. The apolipoprotein E polymorphism in Greenland Inuit in its global perspective. Hum. Genet. 1996, 98, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Bagyinszky, E.; An, S.S.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Crean, S.; Mercaldi, C.J.; Collins, J.M.; Boyd, D.; Cook, M.N.; Arrighi, H.M. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: A systematic review and meta-analysis. Neuroepidemiology 2012, 38, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.D.; Khaleeq, A.; Chan, W.; Pavlik, V.N.; Doody, R.S. Apolipoprotein E polymorphism and age at onset of Alzheimer’s disease in a quadriethnic sample. Dement. Geriatr. Cogn. Disord. 2010, 30, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Sando, S.B.; Melquist, S.; Cannon, A.; Hutton, M.L.; Sletvold, O.; Saltvedt, I.; White, L.R.; Lydersen, S.; Aasly, J.O. APOE epsilon 4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from central Norway. BMC Neurol. 2008, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; PericakVance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease—A meta-analysis. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C. Mild cognitive impairment: Transition between aging and Alzheimer’s disease. Neurologia 2000, 15, 93–101. [Google Scholar] [CrossRef]
- Ma, F.; Wang, J. Apolipoprotein ε4-allele as a significant risk factor for conversion from mild cognitive impairment to Alzheimer’s disease: A meta-analysis of prospective studies. J. Mol. Neurosci. 2013, 50, 257–263. [Google Scholar] [CrossRef]
- Elias-Sonnenschein, L.S.; Wolfgang, V.; Ramakers, I.H.G.B.; Verhey, F.R.J.; Pieter Jelle, V. Predictive value of APOE-ε4 allele for progression from MCI to AD-type dementia: A meta-analysis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, D.A.; Taylor, S.L.; Fullerton, S.M.; Weiss, K.M.; Clark, A.G.; Stengard, J.H.; Salomaa, V.; Boerwinkle, E.; Sing, C.F. Sequence diversity and large-scale typing of SNPs in the human apolipoprotein E gene. Genome Res. 2000, 10, 1532–1545. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Thomas, G.; de Zulueta, M.P.; De Gennes, J.L.; Thomas, M.; Cassaigne, A.; Bereziat, G.; Iron, A. Common and rare genotypes of human apolipoprotein E determined by specific restriction profiles of polymerase chain reaction-amplified DNA. Clin. Chem. 1994, 40, 24–29. [Google Scholar] [PubMed]
- Yamamura, T.; Dong, L.M.; Yamamoto, A. Characterization of apolipoprotein E7 (Glu(244)-->Lys, Glu(245)--->Lys), a mutant apolipoprotein E associated with hyperlipidemia and atherosclerosis. J. Lipid Res. 1999, 40, 253–259. [Google Scholar] [PubMed]
- Youn, Y.C.; Lim, Y.K.; Han, S.-H.; Giau, V.V.; Lee, M.-K.; Park, K.-Y.; Kim, S.; Bagyinszky, E.; An, S.S.A.; Kim, H.R. Apolipoprotein ε7 allele in memory complaints: Insights through protein structure prediction. Clin. Interv. Aging 2017, 12, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Bizzarro, A.; Seripa, D.; Acciarri, A.; Matera, M.G.; Pilotto, A.; Tiziano, F.D.; Brahe, C.; Masullo, C. The complex interaction between APOE promoter and AD: An Italian case-control study. Eur. J. Hum. Genet. 2009, 17, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Laws, S.M.; Hone, E.; Gandy, S.; Martins, R.N. Expanding the association between the APOE gene and the risk of Alzheimer’s disease: Possible roles for APOE promoter polymorphisms and alterations in APOE transcription. J. Neurochem. 2003, 84, 1215–1236. [Google Scholar] [CrossRef] [PubMed]
- Artiga, M.J.; Bullido, M.J.; Sastre, I.; Recuero, M.; Garcia, M.A.; Aldudo, J.; Vazquez, J.; Valdivieso, F. Allelic polymorphisms in the transcriptional regulatory region of apolipoprotein E gene. FEBS Lett 1998, 421, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Xin, X.Y.; Ding, J.Q.; Chen, S.D. Apolipoprotein E promoter polymorphisms and risk of Alzheimer’s disease: Evidence from meta-analysis. J. Alzheimers Dis. 2010, 19, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Gao, Y.; Liu, L.; Li, Y. Association between polymorphisms in the promoter region of the apolipoprotein E (APOE) gene and Alzheimer’s disease: A meta-analysis. EXCLI J 2017, 16, 921–938. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.R.; He, Y.; Tekin, S.; Xu, J.; Xu, J.; Charles, H.C. Impact of APOE in mild cognitive impairment. Neurology 2004, 63, 1898–1901. [Google Scholar] [CrossRef] [PubMed]
- El Haj, M.; Antoine, P.; Amouyel, P.; Lambert, J.C.; Pasquier, F.; Kapogiannis, D. Apolipoprotein E (APOE) epsilon4 and episodic memory decline in Alzheimer’s disease: A review. Ageing Res. Rev. 2016, 27, 15–22. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.C.; Murphy, S.E.; Zamboni, G.; Nobre, A.C.; Mackay, C.E. APOE genotype and cognition in healthy individuals at risk of Alzheimer’s disease: A review. Cortex 2018, 104, 103–123. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Van der Vlies, A.E.; Pijnenburg, Y.A.; Koene, T.; Klein, M.; Kok, A.; Scheltens, P.; van der Flier, W.M. Cognitive impairment in Alzheimer’s disease is modified by APOE genotype. Dement. Geriatr. Cogn. Disord. 2007, 24, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Snowden, J.S.; Stopford, C.L.; Julien, C.L.; Thompson, J.C.; Davidson, Y.; Gibbons, L.; Pritchard, A.; Lendon, C.L.; Richardson, A.M.; Varma, A. Cognitive Phenotypes in Alzheimer’s Disease and Genetic Risk. Cortex 2007, 43, 835–845. [Google Scholar] [CrossRef]
- Wolk, D.A.; Dickerson, B.C. Apolipoprotein E (APOE) genotype has dissociable effects on memory and attentional-executive network function in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 10256–10261. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, S.; Yoo, H.; Jang, H.; Kim, Y.; Kim, K.W.; Jang, Y.K.; Lee, J.S.; Kim, S.T.; Kim, S.; et al. The Impact of APOE varepsilon4 in Alzheimer’s Disease Differs According to Age. J. Alzheimers Dis. 2018, 61, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, S.; Scarmeas, N.; Helzner, E.; Glymour, M.M.; Brandt, J.; Albert, M.; Blacker, D.; Stern, Y. APOE epsilon 4 allele predicts faster cognitive decline in mild Alzheimer disease. Neurology 2008, 70, 1842–1849. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.L.; Fennema-Notestine, C.; Holland, D.; McEvoy, L.K.; Stricker, N.H.; Salmon, D.P.; Dale, A.M.; Bondi, M.W.; Alzheimer’s Disease Neuroimaging Initiative. APOE interacts with age to modify rate of decline in cognitive and brain changes in Alzheimer’s disease. Alzheimers Dement. 2014, 10, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Ramakers, I.H.; Visser, P.J.; Aalten, P.; Bekers, O.; Sleegers, K.; van Broeckhoven, C.L.; Jolles, J.; Verhey, F.R. The association between APOE genotype and memory dysfunction in subjects with mild cognitive impairment is related to age and Alzheimer pathology. Dement. Geriatr. Cogn. Disord. 2008, 26, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Whitehair, D.C.; Sherzai, A.; Emond, J.; Raman, R.; Aisen, P.S.; Petersen, R.C.; Fleisher, A.S.; Alzheimer’s Disease Neuroimaging Initiative. Influence of apolipoprotein E varepsilon4 on rates of cognitive and functional decline in mild cognitive impairment. Alzheimers Dement. 2010, 6, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.K.; Albrecht, M.A.; Greg, S.; Lautenschlager, N.T.; Ellis, K.A.; Paul, M.; Cassandra, S.; Kevin, T.; Ralph, M.; Masters, C.L. Lack of reliable evidence for a distinctive ε4-related cognitive phenotype that is independent from clinical diagnostic status: Findings from the Australian Imaging, Biomarkers and Lifestyle Study. Brain 2013, 136, 2201–2216. [Google Scholar] [CrossRef] [PubMed]
- Lyall, D.M.; Ward, J.; Ritchie, S.J.; Davies, G.; Cullen, B.; Celis, C.; Bailey, M.E.; Anderson, J.; Evans, J.; McKay, D.F.; et al. Alzheimer disease genetic risk factor APOE e4 and cognitive abilities in 111,739 UK Biobank participants. Age Ageing 2016, 45, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, N.M.; Callahan, J.L.; Hawkins, K.A. The effects of apolipoprotein E on non-impaired cognitive functioning: A meta-analysis. Neurobiol. Aging 2011, 32, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.R.; Lyons, M.J.; Franz, C.E.; Grant, M.D.; Boake, C.; Jacobson, K.C.; Xian, H.; Schellenberg, G.D.; Eisen, S.A.; Kremen, W.S. Apolipoprotein E genotype and memory in the sixth decade of life. Neurology 2008, 70, 1771–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Pardo, L.M.; Schuur, M.; Sanchez-Juan, P.; Isaacs, A.; Sleegers, K.; de Koning, I.; Zorkoltseva, I.V.; Axenovich, T.I.; Witteman, J.C.; et al. The apolipoprotein E gene and its age-specific effects on cognitive function. Neurobiol. Aging 2010, 31, 1831–1833. [Google Scholar] [CrossRef] [PubMed]
- Kerchner, G.A.; Berdnik, D.; Shen, J.C.; Bernstein, J.D.; Fenesy, M.C.; Deutsch, G.K.; Wyss-Coray, T.; Rutt, B.K. APOE epsilon4 worsens hippocampal CA1 apical neuropil atrophy and episodic memory. Neurology 2014, 82, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Espeseth, T.; Westlye, L.T.; Fjell, A.M.; Walhovd, K.B.; Rootwelt, H.; Reinvang, I. Accelerated age-related cortical thinning in healthy carriers of apolipoprotein E ε4. Neurobiol. Aging 2008, 29, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Luciano, M.; Gow, A.J.; Harris, S.E.; Hayward, C.; Allerhand, M.; Starr, J.M.; Visscher, P.M.; Deary, I.J. Cognitive ability at age 11 and 70 years, information processing speed, and APOE variation: The Lothian Birth Cohort 1936 study. Psychol. Aging 2009, 24, 129. [Google Scholar] [CrossRef] [PubMed]
- Izaks, G.J.; Gansevoort, R.T.; van der Knaap, A.M.; Navis, G.; Dullaart , R.P.F.; Slaets, J.P.J. The association of APOE genotype with cognitive function in persons aged 35 years or older. PLoS ONE 2011, 6, e27415. [Google Scholar] [CrossRef] [PubMed]
- Quintino-Santos, S.; Diniz, B.S.; Firmo, J.O.; Moriguchi, E.H.; Lima-Costa, M.F.; Castro-Costa, E. APOE epsilon4 allele is associated with worse performance in memory dimensions of the mini-mental state examination: The Bambui Cohort Study of Aging. Int. J. Geriatr. Psychiatry 2015, 30, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Caselli, R.J.; Dueck, A.C.; Osborne, D.; Sabbagh, M.N.; Connor, D.J.; Ahern, G.L.; Baxter, L.C.; Rapcsak, S.Z.; Shi, J.; Woodruff, B.K.; et al. Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. N. Engl. J. Med. 2009, 361, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Caselli, R.J.; Dueck, A.C.; Locke, D.E.; Hoffman-Snyder, C.R.; Woodruff, B.K.; Rapcsak, S.Z.; Reiman, E.M. Longitudinal modeling of frontal cognition in APOE epsilon4 homozygotes, heterozygotes, and noncarriers. Neurology 2011, 76, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Schiepers, O.J.; Harris, S.E.; Gow, A.J.; Pattie, A.; Brett, C.E.; Starr, J.M.; Deary, I.J. APOE E4 status predicts age-related cognitive decline in the ninth decade: Longitudinal follow-up of the Lothian Birth Cohort 1921. Mol. Psychiatry 2012, 17, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Rawle, M.J.; Davis, D.; Bendayan, R.; Wong, A.; Kuh, D.; Richards, M. Apolipoprotein-E (Apoe) epsilon4 and cognitive decline over the adult life course. Transl. Psychiatry 2018, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Barnes, L.L.; Arvanitakis, Z.; Yu, L.; Kelly, J.; De Jager, P.L.; Bennett, D.A. Apolipoprotein E and change in episodic memory in blacks and whites. Neuroepidemiology 2013, 40, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Hendrie, H.C.; Murrell, J.; Baiyewu, O.; Lane, K.A.; Purnell, C.; Ogunniyi, A.; Unverzagt, F.W.; Hall, K.; Callahan, C.M.; Saykin, A.J.; et al. APOE epsilon4 and the risk for Alzheimer disease and cognitive decline in African Americans and Yoruba. Int. Psychogeriatr. 2014, 26, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Lipnicki, D.M.; Crawford, J.D.; Dutta, R.; Thalamuthu, A.; Kochan, N.A.; Andrews, G.; Lima-Costa, M.F.; Castro-Costa, E.; Brayne, C.; Matthews, F.E.; et al. Age-related cognitive decline and associations with sex, education and apolipoprotein E genotype across ethnocultural groups and geographic regions: A collaborative cohort study. PLoS Med. 2017, 14, e1002261. [Google Scholar] [CrossRef] [PubMed]
- Tuminello, E.R.; Han, S.D. The apolipoprotein e antagonistic pleiotropy hypothesis: Review and recommendations. Int. J. Alzheimers Dis. 2011, 2011, 726197. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, C.; Tabet, N.; Rusted, J. The APOE paradox: Do attentional control differences in mid-adulthood reflect risk of late-life cognitive decline. Neurobiol. Aging 2016, 48, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Shu, N.; Li, X.; Ma, C.; Zhang, J.; Chen, K.; Liang, Y.; Chen, Y.; Zhang, Z. Effects of APOE promoter polymorphism on the topological organization of brain structural connectome in nondemented elderly. Hum. Brain Mapp. 2015, 36, 4847–4858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, P.; Li, X.; Ma, C.; Zhang, S.; Liu, Z.; Chen, K.; Ai, L.; Chang, J.; Zhang, Z. The Effects of an APOE Promoter Polymorphism on Human White Matter Connectivity during Non-Demented Aging. J. Alzheimers Dis. 2017, 55, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, P.; Gu, B.; Liu, Z.; Li, X.; Evans, A.C.; Gong, G.; Zhang, Z. The effects of an APOE promoter polymorphism on human cortical morphology during nondemented aging. J. Neurosci. 2015, 35, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhang, Y.; Li, X.; Zhang, J.; Chen, K.; Liang, Y.; Chen, Y.; Liu, Z.; Zhang, Z. Is there a significant interaction effect between apolipoprotein E rs405509 T/T and epsilon4 genotypes on cognitive impairment and gray matter volume? Eur. J. Neurol. 2016, 23, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Rantalainen, V.; Lahti, J.; Henriksson, M.; Kajantie, E.; Tienari, P.; Eriksson, J.G.; Raikkonen, K. APOE and aging-related cognitive change in a longitudinal cohort of men. Neurobiol. Aging 2016, 44, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, M.F.; Martin, X.E.; Alcelay, L.G.; Flores, J.C.; Valiente, J.M.; Juanbeltz, B.I.; Beldarrain, M.A.; Lopez, J.M.; Gonzalez-Fernandez, M.C.; Salazar, A.M.; et al. The COMT Val158 Met polymorphism as an associated risk factor for Alzheimer disease and mild cognitive impairment in APOE 4 carriers. BMC Neurosci. 2009, 10, 125. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.D.; Summers, M.J.; Saunders, N.L.; Janssen, P.; Stuart, K.E.; Vickers, J.C. APOE and BDNF Val66Met polymorphisms combine to influence episodic memory function in older adults. Behav. Brain Res. 2014, 271, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Gomar, J.J.; Conejero-Goldberg, C.; Huey, E.D.; Davies, P.; Goldberg, T.E.; Alzheimer’s Disease Neuroimaging Initiative. Lack of neural compensatory mechanisms of BDNF val66met met carriers and APOE E4 carriers in healthy aging, mild cognitive impairment, and Alzheimer’s disease. Neurobiol. Aging 2016, 39, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Persson, N.; Lavebratt, C.; Wahlin, A. Synergy effects of HbA1c and variants of APOE and BDNFVal66Met explains individual differences in memory performance. Neurobiol. Learn. Mem. 2013, 106, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Thow, M.E.; Summers, M.J.; Summers, J.J.; Saunders, N.L.; Vickers, J.C. Variations in the APOE allele or BDNF Val66Met polymorphism are not associated with changes in cognitive function following a tertiary education intervention in older adults: The Tasmanian Healthy Brain Project. Neurobiol. Aging 2017, 55, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.N.; Liu, H.C.; Liu, T.Y.; Chu, A.; Hong, C.J.; Lin, K.N.; Chi, C.W. Estrogen-metabolizing gene COMT polymorphism synergistic APOE epsilon4 allele increases the risk of Alzheimer disease. Dement. Geriatr. Cogn. Disord. 2005, 19, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, S.; Backman, L.; Dixon, R.A. Executive function performance and change in aging is predicted by apolipoprotein E, intensified by catechol-O-methyltransferase and brain-derived neurotrophic factor, and moderated by age and lifestyle. Neurobiol. Aging 2017, 52, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-E.; Seltman, H.; Peskind, E.R.; Galloway, N.; Zhou, P.X.; Rosenthal, E.; Wijsman, E.M.; Tsuang, D.W.; Devlin, B.; Schellenberg, G.D. Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer’s disease: Patterns of linkage disequilibrium and disease/marker association. Genomics 2007, 89, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Roses, A.D.; Lutz, M.W.; Amrine-Madsen, H.; Saunders, A.M.; Crenshaw, D.G.; Sundseth, S.S.; Huentelman, M.J.; Welsh-Bohmer, K.A.; Reiman, E.M. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenom. J. 2009, 10, 375. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.W.; Crenshaw, D.; Welsh-Bohmer, K.A.; Burns, D.K.; Roses, A.D. New Genetic Approaches to AD: Lessons from APOE-TOMM40 Phylogenetics. Curr. Neurol. Neurosci. Rep. 2016, 16, 48. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Rue, A.L.; Hermann, B.P.; Xu, G.; Koscik, R.L.; Jonaitis, E.M.; Bendlin, B.B.; Hogan, K.J.; Roses, A.D.; Saunders, A.M. The effect of TOMM40 poly-T length on gray matter volume and cognition in middle-aged persons with APOE ε3/ε3 genotype. Alzheimers Dement. 2011, 7, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Lutz, M.W.; Wilson, R.S.; Burns, D.K.; Roses, A.D.; Saunders, A.M.; Gaiteri, C.; De Jager, P.L.; Barnes, L.L.; Bennett, D.A. TOMM40’523 variant and cognitive decline in older persons with APOE epsilon3/3 genotype. Neurology 2017, 88, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Louwersheimer, E.; Cohn-Hokke, P.E.; Pijnenburg, Y.A.; Weiss, M.M.; Sistermans, E.A.; Rozemuller, A.J.; Hulsman, M.; van Swieten, J.C.; van Duijn, C.M.; Barkhof, F.; et al. Rare Genetic Variant in SORL1 May Increase Penetrance of Alzheimer’s Disease in a Family with Several Generations of APOE-varepsilon4 Homozygosity. J. Alzheimers Dis. 2017, 56, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Barral, S.; Bird, T.; Goate, A.; Farlow, M.R.; Diaz-Arrastia, R.; Bennett, D.A.; Graff-Radford, N.; Boeve, B.F.; Sweet, R.A.; Stern, Y.; et al. Genotype patterns at PICALM, CR1, BIN1, CLU, and APOE genes are associated with episodic memory. Neurology 2012, 78, 1464–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharesouran, J.; Rezazadeh, M.; Khorrami, A.; Ghojazadeh, M.; Talebi, M. Genetic evidence for the involvement of variants at APOE, BIN1, CR1, and PICALM loci in risk of late-onset Alzheimer’s disease and evaluation for interactions with APOE genotypes. J. Mol. Neurosci. 2014, 54, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Keenan, B.T.; Shulman, J.M.; Chibnik, L.B.; Raj, T.; Tran, D.; Sabuncu, M.R.; Alzheimer’s Disease Neuroimaging Initiative; Allen, A.N.; Corneveaux, J.J.; Hardy, J.A.; et al. A coding variant in CR1 interacts with APOE-epsilon4 to influence cognitive decline. Hum. Mol. Genet. 2012, 21, 2377–2388. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.C.; Lee, W.J.; Hwang, J.P.; Wang, Y.F.; Tsai, C.F.; Wang, P.N.; Wang, S.J.; Fuh, J.L. ABCA7 gene and the risk of Alzheimer’s disease in Han Chinese in Taiwan. Neurobiol. Aging 2014, 35, 2423. [Google Scholar] [CrossRef] [PubMed]
- Casati, M.; Ferri, E.; Gussago, C.; Mazzola, P.; Abbate, C.; Bellelli, G.; Mari, D.; Cesari, M.; Arosio, B. Increased expression of TREM2 in peripheral cells from mild cognitive impairment patients who progress into Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Jendresen, C.; Arskog, V.; Daws, M.R.; Nilsson, L.N.G. The Alzheimer’s disease risk factors apolipoprotein E and TREM2 are linked in a receptor signaling pathway. J. Neuroinflamm. 2017, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Sleegers, K.; Bettens, K.; De Roeck, A.; Van Cauwenberghe, C.; Cuyvers, E.; Verheijen, J.; Struyfs, H.; Van Dongen, J.; Vermeulen, S.; Engelborghs, S.; et al. A 22-single nucleotide polymorphism Alzheimer’s disease risk score correlates with family history, onset age, and cerebrospinal fluid Abeta42. Alzheimers Dement. 2015, 11, 1452–1460. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rodríguez, E.; Sánchez-Juan, P.; Vázquez-Higuera, J.L.; Mateo, I.; Pozueta, A.; Berciano, J.; Cervantes, S.; Alcolea, D.; Martinez-Lage, P.; Clarimón, J. Genetic risk score predicting accelerated progression from mild cognitive impairment to Alzheimer’s disease. J. Neural Transm. 2013, 120, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Verhaaren, B.F.J.; Vernooij, M.W.; Koudstaal, P.J.; Uitterlinden, A.G.; Duijn, C.M.V.; Hofman, A.; Breteler, M.M.B.; Ikram, M.A. Alzheimer’s Disease Genes and Cognition in the Nondemented General Population. Biol. Psychiatry 2013, 73, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Marden, J.R.; Walter, S.; Tchetgen Tchetgen, E.J.; Kawachi, I.; Glymour, M.M. Validation of a polygenic risk score for dementia in black and white individuals. Brain Behav. 2014, 4, 687–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marden, J.R.; Mayeda, E.R.; Walter, S.; Vivot, A.; Tchetgen, E.J.T.; Kawachi, I.; Glymour, M.M. Using an Alzheimer Disease Polygenic Risk Score to Predict Memory Decline in Black and White Americans Over 14 Years of Follow-up. Alzheimer Dis. Assoc. Disord. 2016, 30, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Qianyi, X.; Zhi-Jun, L.; Sha, T.; Yi-Min, S.; Deke, J.; Hong-Lei, L.; Haitao, C.; Xu, L.; Brittany, L.; Chi-Hsiung, W. Risk prediction for sporadic Alzheimer’s disease using genetic risk score in the Han Chinese population. Oncotarget 2015, 6, 36955–36964. [Google Scholar]
- Lee, J.J.; Kang, S.; Lee, W.; Gunasekaran, T.I.; Gim, J.; Wi, S.O.; Kang, D.O.; Choo, I.; Kim, B.C.; Kim, H.; et al. Risk prediction of alzheimer’s disease with age stratification using polygenic risk scores. Alzheimers Dement. 2018, 14, P339–P340. [Google Scholar] [CrossRef]
- Lacour, A.; Espinosa, A.; Louwersheimer, E.; Heilmann, S.; Hernández, I.; Wolfsgruber, S.; Fernández, V.; Wagner, H.; Rosende-Roca, M.; Mauleón, A.; et al. Genome-wide significant risk factors for Alzheimer’s disease: Role in progression to dementia due to Alzheimer’s disease among subjects with mild cognitive impairment. Mol. Psychiatry 2017, 22, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Louwersheimer, E.; Wolfsgruber, S.; Espinosa, A.; Lacour, A.; Heilmann-Heimbach, S.; Alegret, M.; Hernandez, I.; Rosende-Roca, M.; Tarraga, L.; Boada, M.; et al. Alzheimer’s disease risk variants modulate endophenotypes in mild cognitive impairment. Alzheimers Dement. 2016, 12, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Das, D.; Cherbuin, N.; Anstey, K.J.; Easteal, S. Association of genetic risk factors with cognitive decline: The PATH through life project. Neurobiol. Aging 2016, 41, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Carrasquillo, M.M.; Crook, J.E.; Pedraza, O.; Thomas, C.S.; Pankratz, V.S.; Allen, M.; Nguyen, T.; Malphrus, K.G.; Ma, L.; Bisceglio, G.D. Late-onset Alzheimer’s risk variants in memory decline, incident mild cognitive impairment, and Alzheimer’s disease. Neurobiol. Aging 2015, 36, 60–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yu, J.T.; Wang, H.F.; Han, P.R.; Tan, C.C.; Wang, C.; Meng, X.F.; Risacher, S.L.; Saykin, A.J.; Tan, L. APOE genotype and neuroimaging markers of Alzheimer’s disease: Systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2015, 86, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Jansen, W.J.; Rik, O.; Knol, D.L.; Tijms, B.M.; Philip, S.; Verhey, F.R.J.; Pieter Jelle, V.; Pauline, A.; Dag, A.; Daniel, A. Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 2015, 313, 1924–1938. [Google Scholar] [CrossRef] [PubMed]
- Drzezga, A.; Grimmer, T.; Henriksen, G.; Muhlau, M.; Perneczky, R.; Miederer, I.; Praus, C.; Sorg, C.; Wohlschlager, A.; Riemenschneider, M.; et al. Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology 2009, 72, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Grimmer, T.; Tholen, S.; Yousefi, B.H.; Alexopoulos, P.; Forschler, A.; Forstl, H.; Henriksen, G.; Klunk, W.E.; Mathis, C.A.; Perneczky, R.; et al. Progression of cerebral amyloid load is associated with the apolipoprotein E epsilon4 genotype in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Ge, T.; Sabuncu, M.R.; Smoller, J.W.; Sperling, R.A.; Mormino, E.C.; Alzheimer’s Disease Neuroimaging Initiative. Dissociable influences of APOE epsilon4 and polygenic risk of AD dementia on amyloid and cognition. Neurology 2018, 90, e1605–e1612. [Google Scholar] [CrossRef] [PubMed]
- Scheinin, N.M.; Wikman, K.; Jula, A.; Perola, M.; Vahlberg, T.; Rokka, J.; Nagren, K.; Viitanen, M.; Rinne, J.O. Cortical (1)(1)C-PIB uptake is associated with age, APOE genotype, and gender in “healthy aging”. J. Alzheimers Dis. 2014, 41, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, A.S.; Kewei, C.; Xiaofen, L.; Napatkamon, A.; Auttawut, R.; Pradeep, T.; Hillary, P.; Joshi, A.D.; Marwan, S.; Sadowsky, C.H. Apolipoprotein E ε4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol. Aging 2013, 34, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gonneaud, J.; Arenaza-Urquijo, E.M.; Fouquet, M.; Perrotin, A.; Fradin, S.; de La Sayette, V.; Eustache, F.; Chetelat, G. Relative effect of APOE epsilon4 on neuroimaging biomarker changes across the lifespan. Neurology 2016, 87, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Machulda, M.M.; Hagen, C.E.; Christianson, T.J.; Roberts, R.O.; Knopman, D.S.; Vemuri, P.; Lowe, V.J.; Kremers, W.K.; Jack, C.R., Jr.; et al. Influence of amyloid and APOE on cognitive performance in a late middle-aged cohort. Alzheimers Dement. 2016, 12, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarci, K.; Lowe, V.; Przybelski, S.A.; Weigand, S.D.; Senjem, M.L.; Ivnik, R.J.; Preboske, G.M.; Roberts, R.; Geda, Y.E.; Boeve, B.F.; et al. APOE modifies the association between Abeta load and cognition in cognitively normal older adults. Neurology 2012, 78, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Villeneuve, S.; La Joie, R.; Marks, S.M.; Jagust, W.J. Gene-environment interactions: Lifetime cognitive activity, APOE genotype, and beta-amyloid burden. J. Neurosci. 2014, 34, 8612–8617. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tan, L.; Wang, H.F.; Liu, Y.; Hao, X.K.; Tan, C.C.; Jiang, T.; Liu, B.; Zhang, D.Q.; Yu, J.T.; et al. Multiple Effect of APOE Genotype on Clinical and Neuroimaging Biomarkers Across Alzheimer’s Disease Spectrum. Mol. Neurobiol. 2016, 53, 4539–4547. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, P.; Wiste, H.J.; Weigand, S.D.; Knopman, D.S.; Shaw, L.M.; Trojanowski, J.Q.; Aisen, P.S.; Weiner, M.; Petersen, R.C.; Jack, C.R., Jr.; et al. Effect of apolipoprotein E on biomarkers of amyloid load and neuronal pathology in Alzheimer disease. Ann. Neurol. 2010, 67, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Risacher, S.L.; Kim, S.; Shen, L.; Nho, K.; Foroud, T.; Green, R.C.; Petersen, R.C.; Jack, C.R., Jr.; Aisen, P.S.; Koeppe, R.A.; et al. The role of apolipoprotein E (APOE) genotype in early mild cognitive impairment (E-MCI). Front. Aging Neurosci. 2013, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Irina, A.; Arnold, S.E.; Johannes, A.; Beach, T.G.; Bigio, E.H. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Farfel, J.M.; Yu, L.; De Jager, P.L.; Schneider, J.A.; Bennett, D.A. Association of APOE with tau-tangle pathology with and without β-amyloid. Neurobiol. Aging 2015, 37, 19. [Google Scholar] [CrossRef] [PubMed]
- Lautner, R.; Palmqvist, S.; Mattsson, N.; Andreasson, U.; Wallin, A.; Palsson, E.; Jakobsson, J.; Herukka, S.K.; Owenius, R.; Olsson, B.; et al. Apolipoprotein E genotype and the diagnostic accuracy of cerebrospinal fluid biomarkers for Alzheimer disease. JAMA Psychiatry 2014, 71, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.B.; Caselli, R.J.; Reiman, E.M.; Jon, V. APOE and neuroenergetics: An emerging paradigm in Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Caselli, R.J.; Yun, L.S.; Chen, K.; Bandy, D.; Minoshima, S.; Thibodeau, S.N.; Osborne, D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N. Engl. J. Med. 1996, 334, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Kewei, C.; Alexander, G.E.; Caselli, R.J.; Daniel, B.; David, O.; Saunders, A.M.; John, H. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.M.; Chen, K.; Lee, W.; Chen, Y.; Bauer, R.J.; Reiman, E.; Caselli, R.; Bu, G. Peripheral apoE isoform levels in cognitively normal APOE ε3/ε4 individuals are associated with regional gray matter volume and cerebral glucose metabolism. Alzheimers Res. Ther. 2017, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Brandon, J.A.; Farmer, B.C.; Williams, H.C.; Johnson, L.A. APOE and Alzheimer’s Disease: Neuroimaging of Metabolic and Cerebrovascular Dysfunction. Front. Aging Neurosci. 2018, 10, 180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Liu, C.C.; Ingelgom, A.J.V.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Valla, J.; Yaari, R.; Wolf, A.B.; Kusne, Y.; Beach, T.G.; Roher, A.E.; Corneveaux, J.J.; Huentelman, M.J.; Caselli, R.J.; Reiman, E.M. Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE epsilon4 allele, the major late-onset Alzheimer’s susceptibility gene. J. Alzheimers Dis. 2010, 22, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Cherbuin, N.; Leach, L.S.; Christensen, H.; Anstey, K.J. Neuroimaging and APOE genotype: A systematic qualitative review. Dement. Geriatr. Cogn. Disord. 2007, 24, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Saeed, U.; Mirza, S.S.; MacIntosh, B.J.; Herrmann, N.; Keith, J.; Ramirez, J.; Nestor, S.M.; Yu, Q.; Knight, J.; Swardfager, W.; et al. APOE-epsilon4 associates with hippocampal volume, learning, and memory across the spectrum of Alzheimer’s disease and dementia with Lewy bodies. Alzheimers Dement. 2018, 14, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Galve, L.; Lehmann, M.; Hobbs, N.Z.; Clarkson, M.J.; Ridgway, G.R.; Crutch, S.; Ourselin, S.; Schott, J.M.; Fox, N.C.; Barnes, J. Patterns of cortical thickness according to APOE genotype in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2009, 28, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Pievani, M.; Rasser, P.E.; Galluzzi, S.; Benussi, L.; Ghidoni, R.; Sabattoli, F.; Bonetti, M.; Binetti, G.; Thompson, P.M.; Frisoni, G.B. Mapping the effect of APOE epsilon4 on gray matter loss in Alzheimer’s disease in vivo. Neuroimage 2009, 45, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Donix, M.; Burggren, A.C.; Suthana, N.A.; Siddarth, P.; Ekstrom, A.D.; Krupa, A.K.; Jones, M.; Rao, A.; Martin-Harris, L.; Ercoli, L.M.; et al. Longitudinal changes in medial temporal cortical thickness in normal subjects with the APOE-4 polymorphism. Neuroimage 2010, 53, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, S.; Blazey, T.M.; Holtzman, D.M.; Cruchaga, C.; Su, Y.; Morris, J.C.; Benzinger, T.L.S.; Gordon, B.A. Longitudinal brain imaging in preclinical Alzheimer disease: Impact of APOE epsilon4 genotype. Brain 2018, 141, 1828–1839. [Google Scholar] [CrossRef] [PubMed]
- Adamson, M.M.; Landy, K.M.; Duong, S.; Fox-Bosetti, S.; Ashford, J.W.; Murphy, G.M.; Weiner, M.; Taylor, J.L. Apolipoprotein E epsilon4 influences on episodic recall and brain structures in aging pilots. Neurobiol. Aging 2010, 31, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.Y.; Williamson, R.; Laws, S.M.; Villemagne, V.L.; Bourgeat, P.; Fowler, C.; Rainey-Smith, S.; Salvado, O.; Martins, R.N.; Rowe, C.C.; et al. Effect of APOE Genotype on Amyloid Deposition, Brain Volume, and Memory in Cognitively Normal Older Individuals. J. Alzheimers Dis. 2017, 58, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.; Giampietro, V.; Banaschewski, T.; Barker, G.J.; Bokde, A.L.; Buchel, C.; Conrod, P.; Flor, H.; Frouin, V.; Garavan, H.; et al. A Multi-Cohort Study of ApoE varepsilon4 and Amyloid-beta Effects on the Hippocampus in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 56, 1159–1174. [Google Scholar] [CrossRef] [PubMed]
- Habes, M.; Toledo, J.B.; Resnick, S.M.; Doshi, J.; Van der Auwera, S.; Erus, G.; Janowitz, D.; Hegenscheid, K.; Homuth, G.; Volzke, H.; et al. Relationship between APOE Genotype and Structural MRI Measures throughout Adulthood in the Study of Health in Pomerania Population-Based Cohort. AJNR Am. J. Neuroradiol. 2016, 37, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Bunce, D.; Anstey, K.J.; Cherbuin, N.; Gautam, P.; Sachdev, P.; Easteal, S. APOE genotype and entorhinal cortex volume in non-demented community-dwelling adults in midlife and early old age. J. Alzheimers Dis. 2012, 30, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Honea, R.A.; Vidoni, E.; Harsha, A.; Burns, J.M. Impact of APOE on the healthy aging brain: A voxel-based MRI and DTI study. J. Alzheimers Dis. 2009, 18, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Bagepally, B.S.; Halahalli, H.N.; John, J.P.; Kota, L.; Purushottam, M.; Mukherjee, O.; Sivakumar, P.T.; Bharath, S.; Jain, S.; Varghese, M. Apolipoprotein E4 and brain white matter integrity in Alzheimer’s disease: Tract-based spatial statistics study under 3-Tesla MRI. Neurodegener. Dis. 2012, 10, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Jiang, Y.; Wang, Y.; Wu, X.; Ren, J.; Lee, M.S.; Lee, S.; Huang, L. Modulation on brain gray matter activity and white matter integrity by APOE epsilon4 risk gene in cognitively intact elderly: A multimodal neuroimaging study. Behav. Brain Res. 2017, 322, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Verghese, P.B.; Castellano, J.M.; Holtzman, D.M. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011, 10, 241–252. [Google Scholar] [CrossRef]
- Smith, C.D.; Andersen, A.H.; Kryscio, R.J.; Schmitt, F.A.; Kindy, M.S.; Blonder, L.X.; Avison, M.J. Altered brain activation in cognitively intact individuals at high risk for Alzheimer’s disease. Neurology 1999, 53, 1391. [Google Scholar] [CrossRef] [PubMed]
- Trachtenberg, A.J.; Filippini, N.; Mackay, C.E. The effects of APOE-ε4 on the BOLD response. Neurobiol. Aging 2012, 33, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Z.; Zhang, J.; Chen, K.; Yao, L.; Li, X.; Gong, G.; Wang, J.; Zhang, Z. Precuneus degeneration in nondemented elderly individuals with APOE varepsilon4: Evidence from structural and functional MRI analyses. Hum. Brain Mapp. 2017, 38, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, C.C.; Wu, D.; Chi, N.F.; Chen, P.C.; Liao, Y.P.; Chiu, H.W.; Hu, C.J. Effects of the apolipoprotein E epsilon4 allele on functional MRI during n-back working memory tasks in healthy middle-aged adults. AJNR Am. J. Neuroradiol. 2013, 34, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Matura, S.; Prvulovic, D.; Hartmann, D.; Scheibe, M.; Sepanski, B.; Butz, M.; Oertel-Knochel, V.; Knochel, C.; Karakaya, T.; Fusser, F.; et al. Age-Related Effects of the Apolipoprotein E Gene on Brain Function. J. Alzheimers Dis. 2016, 52, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Shi, Y.; Chen, G.; Wang, Z.; Liu, D.; Yue, C.; Ward, B.D.; Li, W.; Xu, Z.; Chen, G.; et al. Opposite Neural Trajectories of Apolipoprotein E 4 and 2 Alleles with Aging Associated with Different Risks of Alzheimer’s Disease. Cereb. Cortex 2016, 26, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Goveas, J.S.; Xie, C.; Chen, G.; Li, W.; Ward, B.D.; Franczak, M.B.; Jones, J.L.; Antuono, P.G.; Li, S.J. Functional network endophenotypes unravel the effects of apolipoprotein E epsilon 4 in middle-aged adults. PLoS ONE 2013, 8, e55902. [Google Scholar] [CrossRef] [PubMed]
- Heise, V.; Filippini, N.; Trachtenberg, A.J.; Suri, S.; Ebmeier, K.P.; Mackay, C.E. Apolipoprotein E genotype, gender and age modulate connectivity of the hippocampus in healthy adults. Neuroimage 2014, 98, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Yuan, Y.; Xie, C.; Bai, F.; You, J.; Li, L.; Li, S.J.; Zhang, Z. Imbalanced hippocampal functional networks associated with remitted geriatric depression and apolipoprotein E epsilon4 allele in nondemented elderly: A preliminary study. J. Affect. Disord. 2014, 164, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Risacher, S.L.; Kim, S.; Nho, K.; Foroud, T.; Shen, L.; Petersen, R.C.; Jack, C.R., Jr.; Beckett, L.A.; Aisen, P.S.; Koeppe, R.A.; et al. APOE effect on Alzheimer’s disease biomarkers in older adults with significant memory concern. Alzheimers Dement. 2015, 11, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Long, H.; Zuo, X.; Yu, C.; Liu, B.; Wang, Z.; Wang, Q.; Wang, F.; Han, Y.; Jia, J. APOE Effects on Default Mode Network in Chinese Cognitive Normal Elderly: Relationship with Clinical Cognitive Performance. PLoS ONE 2015, 10, e0133179. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Xie, C.; Shu, H.; Liao, W.; Wang, Z.; Liu, D.; Zhang, Z. Differential Effects of APOE Genotypes on the Anterior and Posterior Subnetworks of Default Mode Network in Amnestic Mild Cognitive Impairment. J. Alzheimers Dis. 2016, 54, 1409–1423. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Qiu, T.; Jia, Y.; Huang, P.; Xu, X.; Yu, X.; Shen, Z.; Jiaerken, Y.; Guan, X.; Zhou, J.; et al. Intrinsic functional connectivity alterations in cognitively intact elderly APOE epsilon4 carriers measured by eigenvector centrality mapping are related to cognition and CSF biomarkers: A preliminary study. Brain Imaging Behav. 2017, 11, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, K.; Jia, Y.L.; Zeng, Q.; Jiaerken, Y.; Qiu, T.; Huang, P.; Xu, X.; Shen, Z.; Guan, X.; et al. Altered effective connectivity anchored in the posterior cingulate cortex and the medial prefrontal cortex in cognitively intact elderly APOE epsilon4 carriers: A preliminary study. Brain Imaging Behav. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Mann, D.; Goumidi, L.; Harris, J.; Amouyel, P.; Iwatsubo, T.; Lendon, C.; Chartier-Harlin, M.C. Effect of the APOE promoter polymorphisms on cerebral amyloid peptide deposition in Alzheimer’s disease. Lancet 2001, 357, 608–609. [Google Scholar] [CrossRef]
- Pahnke, J.; Walker, L.C.; Schroeder, E.; Vogelgesang, S.; Stausske, D.; Walther, R.; Warzok, R.W. Cerebral beta-amyloid deposition is augmented by the -491AA promoter polymorphism in non-demented elderly individuals bearing the apolipoprotein E epsilon4 allele. Acta Neuropathol. 2003, 105, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, K.A.; Culpan, D.; Kehoe, P.G.; Wilcock, G.K.; Hughes, A.; Love, S. APOE promoter, ACE1 and CYP46 polymorphisms and beta-amyloid in Alzheimer’s disease. Neuroreport 2004, 15, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhang, Y.; Li, X.; Chen, Y.; Zhang, J.; Liu, Z.; Chen, K.; Zhang, Z. The TT allele of rs405509 synergizes with APOE epsilon4 in the impairment of cognition and its underlying default mode network in non-demented elderly. Curr. Alzheimer Res. 2016, 13, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Espeseth, T.; Greenwood, P.M.; Reinvang, I.; Fjell, A.M.; Walhovd, K.B.; Westlye, L.T.; Wehling, E.; Lundervold, A.; Rootwelt, H.; Parasuraman, R. Interactive effects of APOE and CHRNA4 on attention and white matter volume in healthy middle-aged and older adults. Cogn. Affect. Behav. Neurosci. 2006, 6, 31–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgen, K.; Ramirez, A.; Frolich, L.; Tost, H.; Plichta, M.M.; Kolsch, H.; Rakebrandt, F.; Rienhoff, O.; Jessen, F.; Peters, O.; et al. Genetic interaction of PICALM and APOE is associated with brain atrophy and cognitive impairment in Alzheimer’s disease. Alzheimers Dement. 2014, 10, S269–S276. [Google Scholar] [CrossRef] [PubMed]
- Thambisetty, M.; An, Y.; Nalls, M.; Sojkova, J.; Swaminathan, S.; Zhou, Y.; Singleton, A.B.; Wong, D.F.; Ferrucci, L.; Saykin, A.J.; et al. Effect of complement CR1 on brain amyloid burden during aging and its modification by APOE genotype. Biol. Psychiatry 2013, 73, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Dai, X.; Tao, W.; Liu, H.; Li, H.; Yang, C.; Zhang, J.; Li, X.; Chen, Y.; Ma, C.; et al. APOE influences working memory in non-demented elderly through an interaction with SPON1 rs2618516. Hum. Brain Mapp. 2018, 39, 2859–2867. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Qin, W.; Xu, Q.; Xu, L.; Xu, J.; Zhang, P.; Liu, H.; Liu, B.; Jiang, T.; Yu, C. Modulation of APOE and SORL1 genes on hippocampal functional connectivity in healthy young adults. Brain Struct. Funct. 2017, 222, 2877–2889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Liu, H.; Qin, W.; Liu, B.; Jiang, T.; Yu, C. APOE and KIBRA Interactions on Brain Functional Connectivity in Healthy Young Adults. Cereb. Cortex 2017, 27, 4797–4805. [Google Scholar] [CrossRef] [PubMed]
- Porter, T.; Burnham, S.C.; Dore, V.; Savage, G.; Bourgeat, P.; Begemann, K.; Milicic, L.; Ames, D.; Bush, A.I.; Maruff, P.; et al. KIBRA is associated with accelerated cognitive decline and hippocampal atrophy in APOE epsilon4-positive cognitively normal adults with high Abeta-amyloid burden. Sci. Rep. 2018, 8, 2034. [Google Scholar] [CrossRef] [PubMed]
- Martiskainen, H.; Helisalmi, S.; Viswanathan, J.; Kurki, M.; Hall, A.; Herukka, S.K.; Sarajarvi, T.; Natunen, T.; Kurkinen, K.M.; Huovinen, J.; et al. Effects of Alzheimer’s disease-associated risk loci on cerebrospinal fluid biomarkers and disease progression: A polygenic risk score approach. J. Alzheimers Dis. 2015, 43, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Voyle, N.; Patel, H.; Folarin, A.; Newhouse, S.; Johnston, C.; Visser, P.J.; Dobson, R.J.; Kiddle, S.J. Genetic Risk as a Marker of Amyloid-beta and Tau Burden in Cerebrospinal Fluid. J. Alzheimers Dis. 2017, 55, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Fan, C.C.; Mormino, E.C.; Sugrue, L.P.; Broce, I.J.; Hess, C.P.; Dillon, W.P.; Bonham, L.W.; Yokoyama, J.S.; Karch, C.M.; et al. Polygenic hazard score: An enrichment marker for Alzheimer’s associated amyloid and tau deposition. Acta Neuropathol. 2018, 135, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Darst, B.F.; Koscik, R.L.; Racine, A.M.; Oh, J.M.; Krause, R.A.; Carlsson, C.M.; Zetterberg, H.; Blennow, K.; Christian, B.T.; Bendlin, B.B.; et al. Pathway-Specific Polygenic Risk Scores as Predictors of Amyloid-beta Deposition and Cognitive Function in a Sample at Increased Risk for Alzheimer’s Disease. J. Alzheimers Dis. 2017, 55, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Sabuncu, M.R.; Buckner, R.L.; Smoller, J.W.; Lee, P.H.; Fischl, B.; Sperling, R.A.; Alzheimer’s Disease Neuroimaging Initiative. The association between a polygenic Alzheimer score and cortical thickness in clinically normal subjects. Cereb. Cortex 2012, 22, 2653–2661. [Google Scholar] [CrossRef] [PubMed]
- Corlier, F.; Hafzalla, G.; Faskowitz, J.; Kuller, L.H.; Becker, J.T.; Lopez, O.L.; Thompson, P.M.; Braskie, M.N. Systemic inflammation as a predictor of brain aging: Contributions of physical activity, metabolic risk, and genetic risk. Neuroimage 2018, 172, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, X.; Li, A.; Liu, S.; Qin, W.; Yu, C.; Liu, Y.; Liu, B.; Jiang, T. Polygenic risk for Alzheimer’s disease influences precuneal volume in two independent general populations. Neurobiol. Aging 2018, 64, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Harrison, T.M.; Mahmood, Z.; Lau, E.P.; Karacozoff, A.M.; Burggren, A.C.; Small, G.W.; Bookheimer, S.Y. An Alzheimer’s Disease Genetic Risk Score Predicts Longitudinal Thinning of Hippocampal Complex Subregions in Healthy Older Adults. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desikan, R.S.; Fan, C.C.; Wang, Y.; Schork, A.J.; Cabral, H.J.; Cupples, L.A.; Thompson, W.K.; Besser, L.; Kukull, W.A.; Holland, D.; et al. Genetic assessment of age-associated Alzheimer disease risk: Development and validation of a polygenic hazard score. PLoS Med. 2017, 14, e1002258. [Google Scholar] [CrossRef] [PubMed]
- Lupton, M.K.; Strike, L.; Hansell, N.K.; Wen, W.; Mather, K.A.; Armstrong, N.J.; Thalamuthu, A.; McMahon, K.L.; de Zubicaray, G.I.; Assareh, A.A.; et al. The effect of increased genetic risk for Alzheimer’s disease on hippocampal and amygdala volume. Neurobiol. Aging 2016, 40, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Xiao, E.; Chen, Q.; Goldman, A.L.; Tan, H.Y.; Healy, K.; Zoltick, B.; Das, S.; Kolachana, B.; Callicott, J.H.; Dickinson, D.; et al. Late-Onset Alzheimer’s Disease Polygenic Risk Profile Score Predicts Hippocampal Function. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2017, 2, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Foley, S.F.; Tansey, K.E.; Caseras, X.; Lancaster, T.; Bracht, T.; Parker, G.; Hall, J.; Williams, J.; Linden, D.E. Multimodal Brain Imaging Reveals Structural Differences in Alzheimer’s Disease Polygenic Risk Carriers: A Study in Healthy Young Adults. Biol. Psychiatry 2017, 81, 154–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laukka, E.J.; Lövdén, M.; Herlitz, A.; Karlsson, S.; Ferencz, B.; Pantzar, A.; Keller, L.; Graff, C.; Fratiglioni, L.; Bäckman, L. Genetic effects on old-age cognitive functioning: A population-based study. Psychol. Aging 2013, 28, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Ihle, A.; Bunce, D.; Kliegel, M. APOE ε4 and Cognitive Function in Early Life: A meta-analysis. Neuropsychology 2012, 26, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Bathum, L.; Christiansen, L.B.; Vaupel, J.; Mcgue, M.; Christensen, K. Apolipoprotein e genotypes: Relationship to cognitive functioning, cognitive decline, and survival in nonagenarians. J. Am. Geriatr. Soc. 2010, 54, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Østbye, T. Neuropsychological performance in advanced age: Influences of Demographic factors and Apolipoprotein E: Findings from the Cache County Memory Study. Clin. Neuropsychol. 2009, 23, 77–99. [Google Scholar] [Green Version]
- Bunce, D.; Bielak, A.A.; Anstey, K.J.; Cherbuin, N.; Batterham, P.J.; Easteal, S. APOE genotype and cognitive change in young, middle-aged, and older adults living in the community. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Han, S.D.; Bondi, M.W. Revision of the apolipoprotein E compensatory mechanism recruitment hypothesis. Alzheimers Dement. 2008, 4, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Carrion-Baralt, J.R.; Melendez-Cabrero, J.; Rodriguez-Ubinas, H.; Schmeidler, J.; Beeri, M.S.; Angelo, G.; Sano, M.; Silverman, J.M. Impact of APOE epsilon4 on the cognitive performance of a sample of non-demented Puerto Rican nonagenarians. J. Alzheimers Dis. 2009, 18, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Duchek, J.M.; Balota, D.A.; Cortese, M. Prospective memory and apolipoprotein E in healthy aging and early stage Alzheimer’s disease. Neuropsychology 2006, 20, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Bloss, C.S.; Delis, D.C.; Salmon, D.P.; Bondi, M.W. Decreased cognition in children with risk factors for Alzheimer’s disease. Biol. Psychiatry 2008, 64, 904–906. [Google Scholar] [CrossRef] [PubMed]
- Woodard, J.L.; Seidenberg, M.; Nielson, K.A.; Antuono, P.; Guidotti, L.; Durgerian, S.; Zhang, Q.; Lancaster, M.; Hantke, N.; Butts, A. Semantic memory activation in amnestic mild cognitive impairment. Brain 2009, 132, 2068. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Schmitz, T.W.; Trivedi, M.A.; Ries, M.L.; Torgerson, B.M.; Carlsson, C.M.; Asthana, S.; Hermann, B.P.; Sager, M.A. The influence of Alzheimer disease family history and apolipoprotein E epsilon4 on mesial temporal lobe activation. J. Neurosci. 2006, 26, 6069–6076. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.W.; Gentleman, S.M.; Lynch, A.; Graham, D.I. beta A4 amyloid protein deposition in brain after head trauma. Lancet 1991, 338, 1422–1423. [Google Scholar] [CrossRef]
- Teasdale, G.M.; Nicoll, J.A.; Murray, G.; Fiddes, M. Association of apolipoprotein E polymorphism with outcome after head injury. Lancet 1997, 350, 1069–1071. [Google Scholar] [CrossRef]
- Irie, F.; Fitzpatrick, A.; Kuller, O.L.; Peila, R.; Newman, A.; Launer, L. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: The Cardiovascular Health Study Cognition Study. Arch. Neurol. 2008, 65, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Profenno, L.A.; Faraone, S.V. Diabetes and overweight associate with non-APOE4 genotype in an Alzheimer’s disease population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 147B, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Ihab, H.; Farzaneh, S.; Lipsitz, L.A. Apolipoprotein e, carbon dioxide vasoreactivity, and cognition in older adults: Effect of hypertension. J. Am. Geriatr. Soc. 2015, 63, 276–281. [Google Scholar]
- Giri, M.; Zhang, M.; Yang, L. Genes associated with Alzheimer’s disease: An overview and current status. Clin. Interv. Aging 2016, 11, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Kauppi, K.; Fan, C.C.; McEvoy, L.K.; Holland, D.; Tan, C.H.; Chen, C.H.; Andreassen, O.A.; Desikan, R.S.; Dale, A.M.; Alzheimer’s Disease Neuroimaging Initiative. Combining Polygenic Hazard Score With Volumetric MRI and Cognitive Measures Improves Prediction of Progression From Mild Cognitive Impairment to Alzheimer’s Disease. Front. Neurosci. 2018, 12, 260. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.F.; Smith, A.V.; Aspelund, T.; Betensky, R.A.; Smoller, J.W.; Gudnason, V.; Launer, L.J.; Blacker, D. Genetic overlap between vascular pathologies and Alzheimer’s dementia and potential causal mechanisms. Alzheimers Dement 2019, 15, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Suri, S.; Heise, V.; Trachtenberg, A.J.; Mackay, C.E. The forgotten APOE allele: A review of the evidence and suggested mechanisms for the protective effect of APOE ε2. Neurosci. Biobehav. Rev. 2013, 37, 2878–2886. [Google Scholar] [CrossRef] [PubMed]
- Prins, N.D.; van der Flier, W.A.; Knol, D.L.; Fox, N.C.; Brashear, H.R.; Nye, J.S.; Barkhof, F.; Scheltens, P. The effect of galantamine on brain atrophy rate in subjects with mild cognitive impairment is modified by apolipoprotein E genotype: Post-hoc analysis of data from a randomized controlled trial. Alzheimers Res. Ther. 2014, 6, 47. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study | Participants | Genes | Interaction Impact on Disease | Possible Mechanisms Described by the Authors |
|---|---|---|---|---|
| Martinez et al., 2009 [70] | 223 MCI patients, 345 AD and 253 HC | COMT | COMT (Val158 Met) polymorphism is not an independent risk factor for AD or MCI, but shows a synergistic effect with APOE ε4 allele that proves greater in women with AD. | Lowering the estrogen levels of brain. |
| Wang et al., 2005 [75] | 66 AD and 86 HC | COMT | The COMT high-activity genotypes and APOEε4 allele had a synergistic effect on the risk of AD. | A high metabolism of estrogen by COMT may have reduced the protective effect of estrogen in AD. |
| Sapkota et al., 2017 [76] | 634 non-demented older adults | COMT BDNF | APOE ε4+ carriers with BDNF Met/Met genotype and increasing allelic risk in the COMT + BDNF risk panel had poorer executive function performance. | − |
| Ward et al., 2014 [71] | 433 older adults (50–79 years) | BDNF | In BDNF Val homozygotes, the cognitive consequences of APOE polymorphisms were minimal. However, in BDNF Met carriers, the hypothesized beneficial/detrimental effects of APOE polymorphisms were found. | Firstly, there is a biological interaction related to the systems or aging-related roles of the encoded proteins. Secondly, the additive effects of the polymorphisms caused the analyses to reach statistical significance. |
| Gomar et al., 2016 [72] | 175 healthy subjects and 222 with prodromal and established AD | BDNF | BDNF Met and APOE ε4 carriers had thinner posterior cingulate and precuneus cortices in healthy subjects, and longitudinal decline in entorhinal thickness in MCI and AD. | − |
| Persson et al., 2013 [73] | 888 non-demented adults (35–85 years) | BDNF | A joint effect on memory decline in BDNF × APOE × age, with the subjects carrying the Met allele, as well as at least one copy of the APOE ε4 allele showing magnified effect sizes with increasing age on memory decline, while the homozygote Val subjects carrying the ε4 allele showed a decreased slope. | − |
| Yu et al., 2007 [77] | 193 late-onset AD, 232 subjects with no cognitive impairment, and 125 individuals with other neurodegenerative disorders | TOMM40 | It showed intriguing linkage disequilibrium with the ε4 allele and was strongly associated with the risk for developing late onset AD. | − |
| Roses et al., 2009 [78] | 191 AD and 131 HC (mean age: about 75 years) | TOMM40 | Individuals with long poly-T repeats linked to APOE ε3 develop late onset AD on an average of 7 years earlier than individuals with shorter poly-T repeats linked to APOE ε3. | It is possible that the rs10524523 polymorphism, alone or in conjunction with other single-nucleotide polymorphisms in TOMM40, acts at a distance to affect transcription of APOE. |
| Johnson et al., 2011 [80] | 117 healthy APOE ε3 homozygous adults (mean age: about 55 years) | TOMM40 | Those who were homozygous for very long poly-T lengths had poorer memory than those who were homozygous for short poly-T length in APOE ε3/3. | − |
| Yu et al., 2017 [81] | 1151 old people (mean age: about 78.5 years) | TOMM40 | It revealed an association of APOE ε3/3-TOMM40′523 haplotypes with cognitive decline in community-based older persons such that the S/S poly-T genotype is related to faster cognitive decline, primarily in the domains of episodic and semantic memory. | The TOMM40 variant is implicated in affecting the level of neurofilament light proteins in cerebrospinal fluid. |
| Louwersheimer et al., 2017 [82] | A family with 9 AD patients spanning 4 generations, with an inheritance pattern suggestive of autosomal dominant | SORL1 | All four affected family members carried a rare variant in the vacuolar protein sorting domain 10 domain of the SORL1 gene, associated with Aβ protein precursor processing and AD risk. | A combination of homozygous or heterozygous APOEε4 and dysfunctional SORL1 may lead to abnormal increases in extracellular Aβ loads. |
| Barral et al., 2012 [83] | 1365 subjects in the National Institute on Aging Late-Onset Alzheimer’s Disease Family Study | CR1, BIN1, CLU, PICALM | Several genotype patterns influenced episodic memory performance. | − |
| Gharesouran et al., 2014 [84] | 160 patients with late-onset AD and in 163 HC | PICALM, BIN1 | The associations with PICALM and BIN1 were only significant among subjects without the APOE ε4 allele. | − |
| Keenan et al., 2012 [85] | 1709 subjects (697 deceased) from the Religious Orders Study and the Rush Memory and Aging Project | CR1 | A significant interaction between our candidate functional variant rs4844609 and the presence or absence of APOE ε4 on episodic memory decline. | − |
| Liao et al., 2014 [86] | 536 AD cases and 307 cognitive-intact elder controls | ABCA7 | The influence of ABCA7 was only evident in individuals without APOE ε4 alleles but absent in ε4 carriers. | − |
| Casati et al., 2018 [87] | 57 MCI, 50 AD, and 42 non-demented healthy subjects (mean age: about 78.5 years) | TREM2 | Higher TREM2 levels in allele ε4 of apolipoprotein E carriers than non-carriers in MCI and particularly in MCI-AD. | The upregulation of TREM2 could be a mechanism to counteract neuroinflammatory processes in MCI patients who progress to AD. |
| Espeseth et al., 2006 [157] | 230 healthy middle-aged (53–64 years) and older (65–75 years) adults | CHRNA4 | APOE-ε4 carriers who were also CHRNA4 TT homozygotes showed disproportionately slowed reaction time (RT) following invalid location cues. There was also a trend for individuals with combined APOE-ε4/CHRNA4 TT genotypes to show both lower white-matter volume and slower overall RT on the attention task. | It remains for further research to determine which of several underlying mechanisms—acetylcholine synthesis, cholinergic neuronal metabolism, synaptic availability of acetylcholine, the affinity of cholinergic receptors, or other factors—are responsible for the interactive effects of APOE and CHRNA4 on attention. |
| Morgen et al., 2014 [158] | 165 patients with early AD dementia | PICALM | There was a synergistic adverse effect of homozygosity for the PICALM risk allele G in rs3851179 and APOE ε4 on volume in prefrontal and performance on the Trail Making Test. | The APOE and PICALM risk genotypes may contribute to Aβ accumulation through different mechanisms, ultimately leading to synaptic dysfunction and loss. |
| Thambisetty et al., 2013 [159] | 57 non-demented older individuals (mean age: about 78.5 years) and 22 cognitively normal older individuals (mean age: about 77.1 years) | CR1 | Carrying a risk allele of the CR1 rs3818361 results in a reduced brain amyloid burden compared to non-carriers, but only in ε4 non-carriers. | The CR1 risk allele might modify the relationship between APOE genotype and brain amyloid deposition. |
| Liu et al., 2018 [160] | 710 individuals (mean age: about 65 years) | SPON1 | Significant SPON1 × APOE genotype interactions in working memory and executive function performances. The effects of ε4 allele on activation of right inferior frontal gyrus, triangular part were modulated by rs2618516 in a working memory task. | − |
| Shen et al., 2017 [161] | 287 healthy, young, right-handed subjects (mean age: 22.7 ± 2.4 years, ranging from 18 to 29 years) | SORL1 | Significant SORL1 × APOE non-additive interaction was found in negative resting state functional connectivity (rsFC) between the hippocampus and inferior frontal gyrus. Compared with subjects with TT genotype, SORL1 G-allele carriers had a stronger negative rsFC in APOE ε4 carriers, but a weaker negative rsFC in APOE non-ε4 carriers. | − |
| Zhang et al., 2017 [162] | 267 healthy young adults (mean age: about 22.8 years) | KIBRA | Epistatic effects showed APOE × KIBRA interaction in the functional connectivity density (FCD) of the dorsolateral prefrontal cortex (DLPFC). The FCD of the DLPFC showed APOE risk-allele-dependent reduction (ε2 > ε3 > ε4) in KIBRA TT homozygotes, but APOE risk-allele-dependent increase in KIBRA C-carriers. | One candidate explanation for the complex APOE–KIBRA interactions on brain FCD may be the differential effects of genetic variations in APOE and KIBRA on the long-term potentiation of memory-related brain regions. |
| Porter et al., 2018 [163] | 602 CN adults | KIBRA | In comparison to APOE ε4- individuals carrying the rs17070145-T allele, significantly faster rates of cognitive decline, and hippocampal atrophy were observed in individuals who were APOE ε4+ and did not carry the rs17070145-T allele. | Synaptic plasticity, which is altered in AD, is modulated by dendrin, which in turn binds to the protein that KIBRA encodes. |
| Study | Participants | Study Design | SNP | APOE | Conversion Risk | Cognitive Impact | Neuroimaging Impact |
|---|---|---|---|---|---|---|---|
| Sabuncu et al., 2012 [168] | 104 CN (75.9 ± 5.1) and 100 AD (75.1 ± 7.8) | Cross-sectional study | 26 | N | The PGS was significantly associated with CDR-SB, MMSE, and AD diagnosis. | AD-specific cortical thickness was correlated with the PGS, even after controlling for APOE genotype and CSF levels of Aβ42. The association remained significant in CN subjects with levels of CSF Aβ42 in the normal range and in APOE ε3 homozygotes. | |
| Rodriguez-Rodriguez et al., 2013 [90] | 228 MCI | Longitudinal study (26.3 months) | 8 | N | PGS was not associated with risk of conversion from MCI to AD. MCI-converters to AD harboring six or more risk alleles progressed twofold more rapidly to AD when compared with those with less than six risk alleles. | ||
| Verhaaren et al., 2013 [91] | Non-demented 5171 (age range 45–99) | Cross-sectional study | 12 | Y | PGS was primarily associated with memory. | ||
| Marden et al., 2014 [92] | 10401 (memory score sample), 7690 (AD probability scores) non-Hispanic white and black | Cross-sectional study | 10 | Y | Each 0.10 unit change in PGS was associated with larger relative effects on dementia among aged 65+. | Each 0.10 unit change in the PGS was associated with a −0.07 standard deviation difference in memory score among aged 50+. | |
| Carrasquillo et al., 2015 [99] | CN 2674 | Longitudinal study | 10 | Y | PGS was associated with progression to MCI/LOAD. | PGS was associated with worse memory. | |
| Martiskainen et al., 2015 [164] | 890 AD (69.8 ± 8.2) and 701 CN (69.1 ± 6.2) | Cross-sectional study | 22 | Y/N | PGS associated with CSF Aβ42 levels in the clinical cohort, and with soluble Aβ42 levels and γ-secretase activity in the neuropathological cohort. The γ-secretase effect was independent of APOE. | ||
| Xiao et al., 2015 [94] | 459 AD (71.2 ± 9.6), 751 CN (72.7 ± 5.9) Chinese | Cross-sectional study | 3 | N | PGS significantly associated with AD risk. | ||
| Sleegers et al., 2015 [89] | 1162 AD (74.4 ± 8.9) and 1019 CN (76.2 ± 8.5) | Cross-sectional study | 22 | Y | Risk of AD increased with PGS; onset age decreased with increasing PGS. | CSF Aβ42 decreased with increasing PGS. | |
| Andrews et al., 2016 [98] | Non-demented 1689 (62.54 ± 1.51) | Longitudinal study | 12 | Y | PGS was associated with worse performance on episodic memory. | ||
| Harrison et al., 2016 [171] | 66 baseline participants (63.0 ± 10.4) and 45 follow-up participants (63.2±7.8) | Longitudinal study (2 years) | 21 | Y | Both unweighted risk score and weighted risk score correlated strongly with the percentage change in thickness across the whole hippocampal complex, driven by a strong relationship to entorhinal cortex thinning. By contrast, at baseline, the risk scores showed no relationship to thickness in any hippocampal complex subregion. | ||
| Louwersheimer et al., 2016 [97] | 1730 MCI from 4 independent datasets | Longitudinal study | 18 | N | PGS was modestly associated with cognitive decline over time. | PGS was modestly associated with CSF levels of tau and p-tau. | |
| Lupton et al., 2016 [173] | 1674 older (aged >53 years; 17% AD, 39% MCI) and 467 young (16–30 years) adults | Cross-sectional study | Different thresholds | N | PGS associated with reduced hippocampal volume in older CN and MCI. No associations were found in young adults. | ||
| Marden et al., 2016 [93] | 8253 non-Hispanic whites and blacks | Longitudinal study | 22 | Y/N | PGS can predict a more rapid decline in memory in whites and blacks; PGS without APOE ε4 only can predict memory decline in whites. | ||
| Darst et al., 2017 [167] | 1200 at baseline (53.6 ± 6.6) | Longitudinal study | 21 | Y | Non-significant for associations between the PGS and cognitive outcomes. | These additional variants did not add much predictive power over APOE alone on biomarkers of Aβ deposition, neurodegeneration and tau pathology. | |
| Desikan et al., 2017 [172] | More than 80,000 people from two projects | Longitudinal study | 31 | N | ADGC Phase 1: highest PGS quartile, lower age onset and the highest yearly AD incidence rate. APOE ε3/3 individuals: PGS modified expected age of AD onset by more than 10 years between the lowest and highest deciles. Independent cohorts: PGS strongly predicted empirical age of AD onset and longitudinal progression. | PGS was associated with neuropathology (Braak stage of neurofibrillary tangles and Consortium to Establish a Registry for Alzheimer’s Disease score for neurotic plaques) and in vivo markers of AD neurodegeneration (volume loss within the entorhinal cortex and hippocampus) | |
| Foley et al., 2017 [175] | 272 T1 (24.8 ± 6.9), 197 DTI (23.9 ± 5.1), 87 Hopkins Verbal Learning Task (23.9 ± 4.4) | Cross-sectional study | 7 thresholds | Y/N | A significant association between PGS and left hippocampal volume; this effect remained when the APOE gene was excluded. The fractional anisotropy of the right cingulum was inversely correlated with PGS. | ||
| Lacour et al., 2017 [96] | 4 MCI groups 853/812/1245/306 | Longitudinal study | 9 | N | PGS predicted a small effect on the risk of MCI to AD progression in APOE ε4 carriers. | ||
| Voyle et al., 2017 [165] | About 250 people with normal and abnormal CSF Aβ from ADNI | Cross-sectional study | − | N | A case/control PGS is marginally more predictive of Aβ and tau pathology than the basic models (with age, gender and APOE genotype). | ||
| Xiao et al., 2017 [174] | 231 CN (age range 19–55) | Cross-sectional study | 6 thresholds | N | Almost no significant association of PGS with cognition. | There was a significant negative relationship between PGS and hippocampal function. | |
| Ge et al., 2018 [104] | 702 participants (221 CN, 367 MCI, and 114 AD) and a subset of 669 participants | Longitudinal study | Different thresholds | N | Only weak associations between PGS and baseline Aβ were present. PGSs were associated with hippocampal atrophy in Aβ− and weakly associated with baseline hippocampal volume in Aβ+. | ||
| Kauppi et al., 2018 [193] | 336 MCI (baseline age range 55–89) | Longitudinal study (3 year) | 31 | Y | PGS significantly predicted time to progression from MCI to AD over 120 months, and PGS was significantly more predictive than APOE alone. | PGS improved the prediction of change in the CDR-SB score and MMSE over 36 months in MCI at baseline, beyond both APOE and baseline levels of brain atrophy. | |
| Li et al., 2018 [170] | 360 CN (19.4 ± 1.1) in discovery dataset and 323 CN (22.7 ± 2.5) in replication dataset | Cross-sectional study | − | Y/N | No correlation between PGS and any cognitive measure in either sample. | In both cohorts, an elevated PGS was associated with a smaller precuneal volume, and the effect remained after excluding the APOE genotype. | |
| Lin et al., 2019 [194] | 2907 stroke-free individuals (76.73 ± 5.83) | Cross-sectional study | 3 thresholds | Y/N | PGSs were associated with lobar cerebral microbleeds, white-matter lesion load, and coronary artery calcification, mostly explained by single-nucleotide polymorphism in the APOE region. The effect of PGS on cognition was partially but significantly mediated by cerebral microbleeds, white-matter lesions, and coronary artery calcification. | ||
| Tan et al., 2018 [166] | 347 CN (baseline age range 59.7–90.1), 599 MCI (baseline age range 54.4–91.4), and 485 (age at death range = 71.3–108.3) in another cohort | Longitudinal study | 31 | N | Even after accounting for APOE ε4 effects, PGS may be useful in MCI and preclinical AD therapeutic trials to enrich for biomarker-positive individuals at highest risk for short-term clinical progression. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, J.; Tao, W.; Li, X.; Li, H.; Zhang, J.; Wei, D.; Chen, Y.; Zhang, Z. The Contribution of Genetic Factors to Cognitive Impairment and Dementia: Apolipoprotein E Gene, Gene Interactions, and Polygenic Risk. Int. J. Mol. Sci. 2019, 20, 1177. https://doi.org/10.3390/ijms20051177
Fan J, Tao W, Li X, Li H, Zhang J, Wei D, Chen Y, Zhang Z. The Contribution of Genetic Factors to Cognitive Impairment and Dementia: Apolipoprotein E Gene, Gene Interactions, and Polygenic Risk. International Journal of Molecular Sciences. 2019; 20(5):1177. https://doi.org/10.3390/ijms20051177
Chicago/Turabian StyleFan, Jialing, Wuhai Tao, Xin Li, He Li, Junying Zhang, Dongfeng Wei, Yaojing Chen, and Zhanjun Zhang. 2019. "The Contribution of Genetic Factors to Cognitive Impairment and Dementia: Apolipoprotein E Gene, Gene Interactions, and Polygenic Risk" International Journal of Molecular Sciences 20, no. 5: 1177. https://doi.org/10.3390/ijms20051177
APA StyleFan, J., Tao, W., Li, X., Li, H., Zhang, J., Wei, D., Chen, Y., & Zhang, Z. (2019). The Contribution of Genetic Factors to Cognitive Impairment and Dementia: Apolipoprotein E Gene, Gene Interactions, and Polygenic Risk. International Journal of Molecular Sciences, 20(5), 1177. https://doi.org/10.3390/ijms20051177