The Impact of the C-Terminal Region on the Interaction of Topoisomerase II Alpha with Mitotic Chromatin

 , and
, and
Abstract
:1. Introduction
2. Results
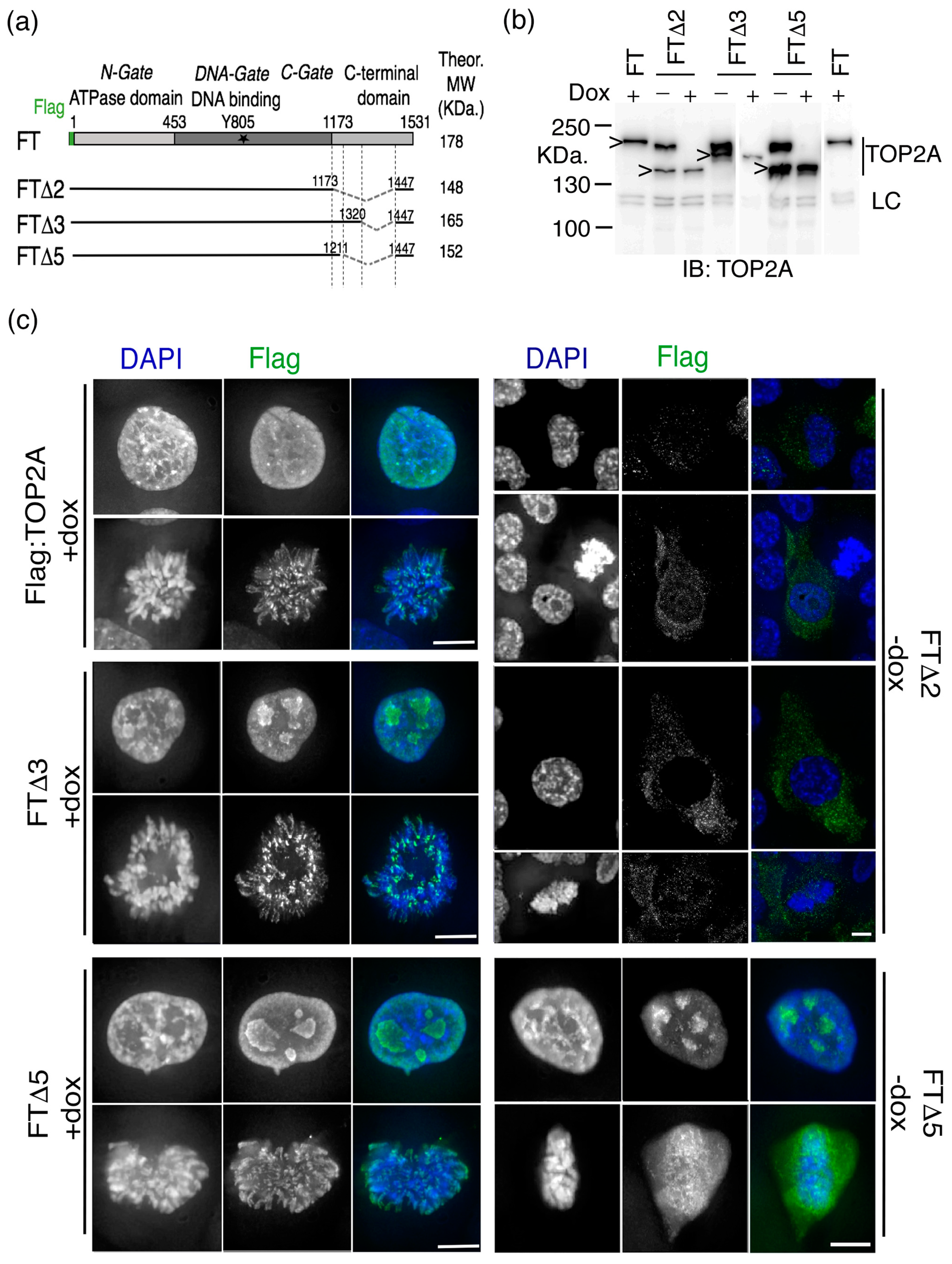
2.1. The Impact of Internal Deletion of the CTD on Localisation of TOP2A to Mitotic Chromatin
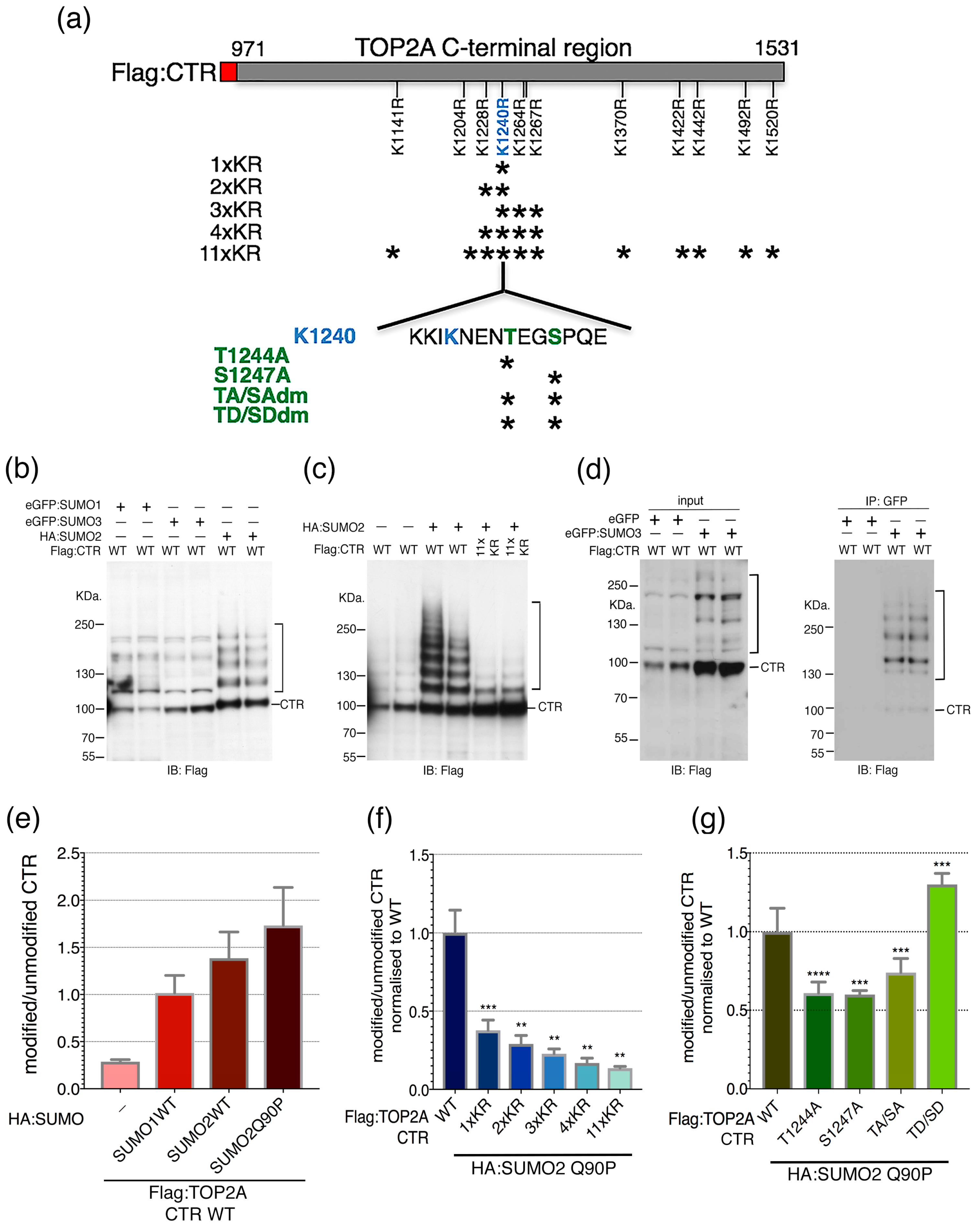
2.2. Candidate SUMOylation Sites within the TOP2A C-Terminal Region (CTR)
2.3. SUMO-Phospho Cross Talk
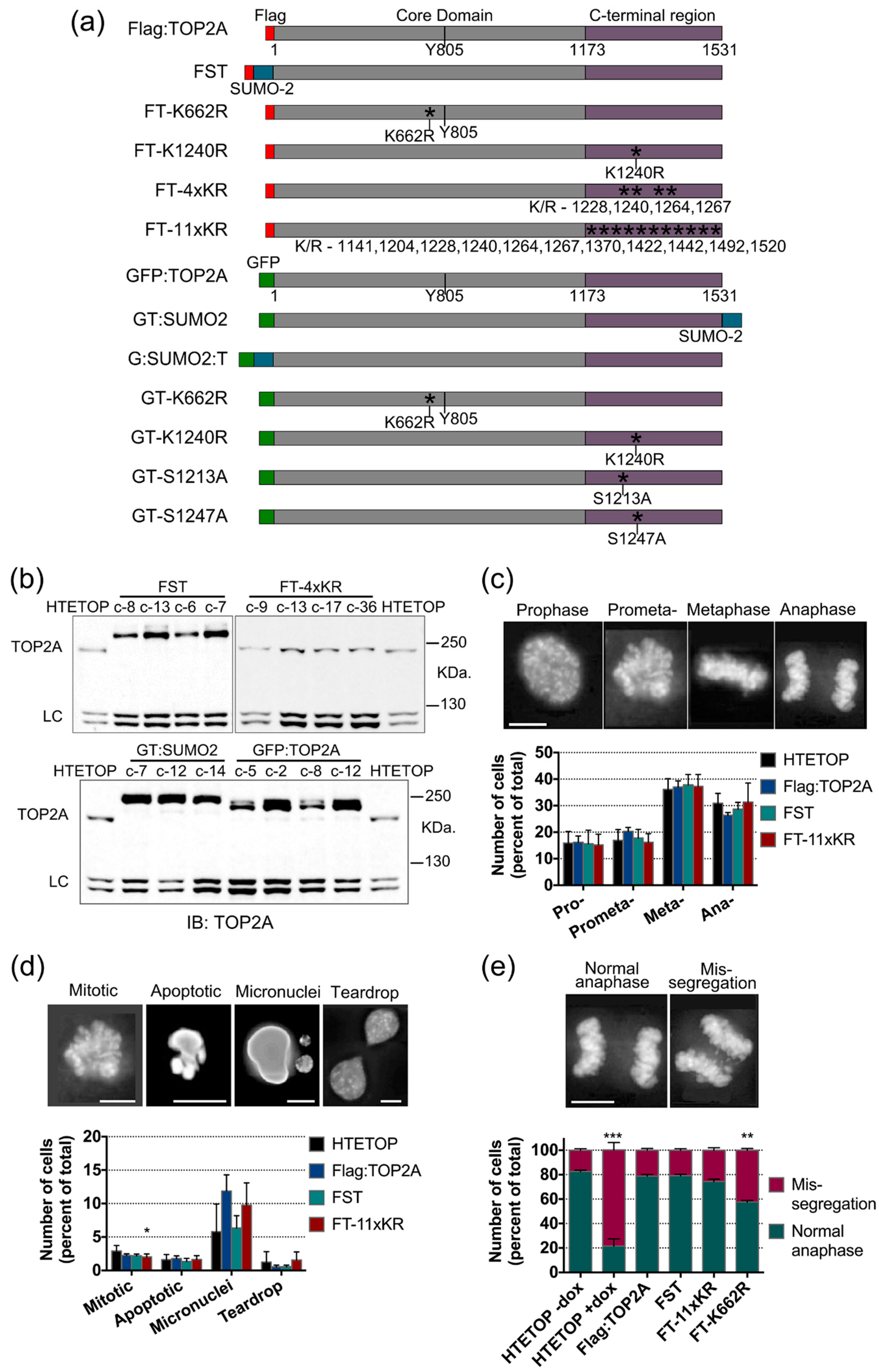
2.4. Mutation of TOP2A SUMO Acceptor Sites and Cell Cycle Progression
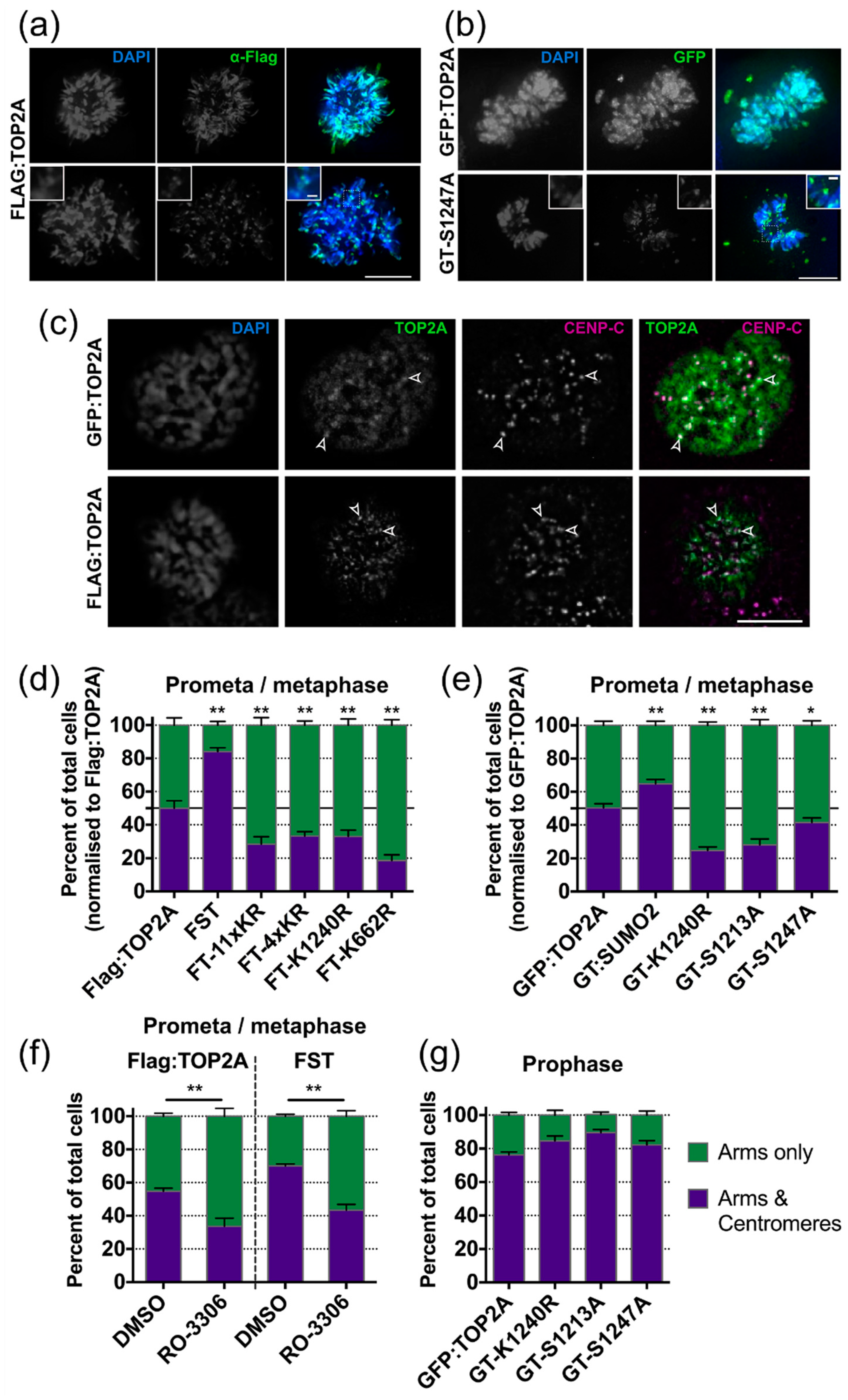
2.5. Factors Influencing TOP2A Localisation in Mitosis
2.5.1. SUMOylation
2.5.2. Phosphorylation
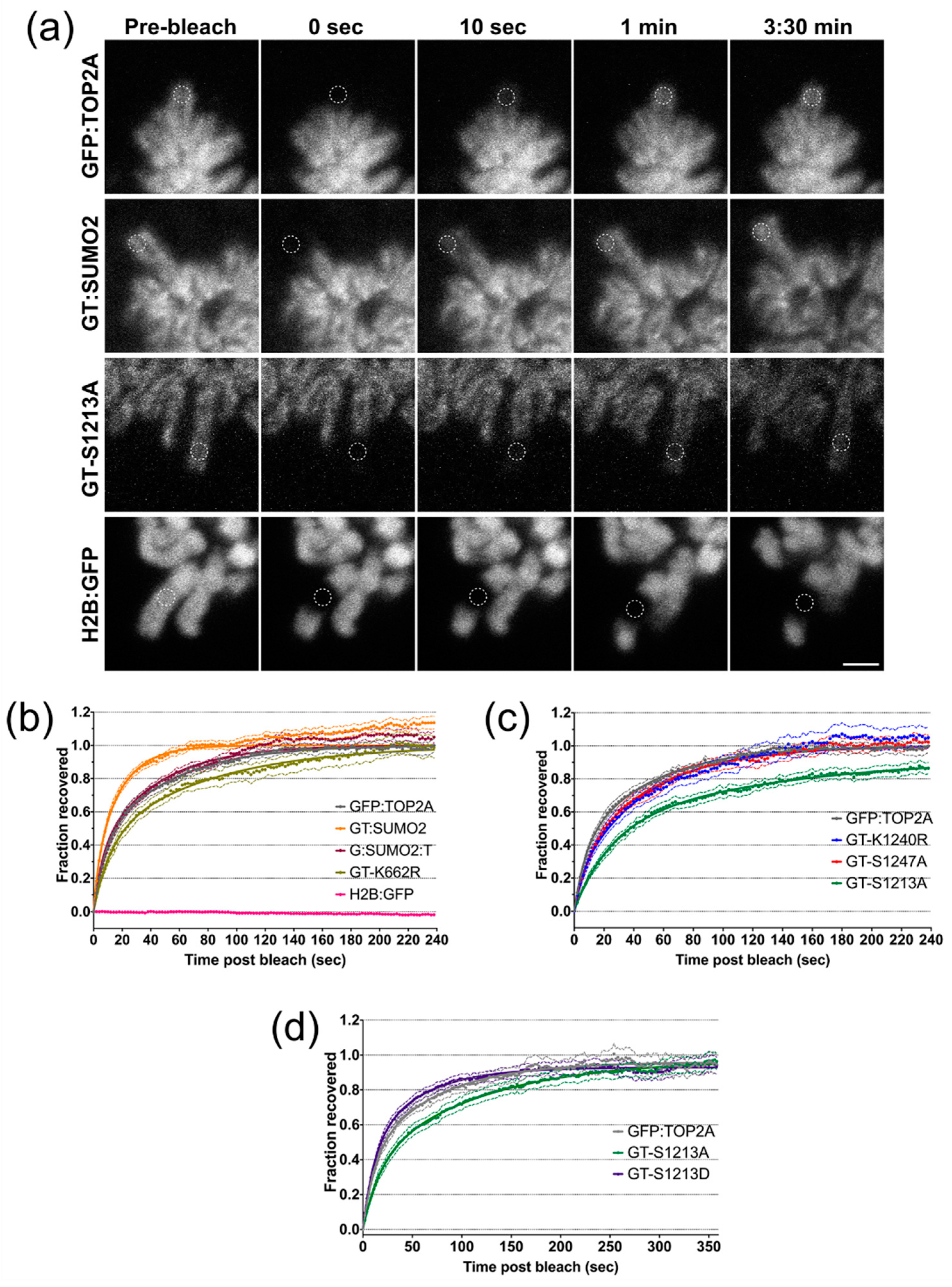
2.6. Factors Influencing the Dynamic Exchange of TOP2A on Mitotic Chromatin
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Molecular Biology
4.3. Protein Work
4.3.1. Western Blot Analysis
4.3.2. Immunoprecipitation
4.4. Microscopy
4.4.1. Assessment of Mitosis and Nuclear Phenotypes
4.4.2. Indirect Immunofluorescence
4.4.3. Direct GFP Visualisation
4.4.4. Fluorescence Recovery after Photobleaching (FRAP)
4.5. Cell culture
4.5.1. Cell Lines
4.5.2. Stable Transfections
4.5.3. Transient Transfections
4.5.4. Drug Treatments
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ChT | Chromatin tether |
| CTD | C-Terminal Domain |
| CTR | C-terminal region |
| DMSO | dimethyl sulphoxide |
| GFP | enhanced Green Fluorescent Protein |
| HMW | High Molecular Weight |
| KDa | KiloDalton |
| NLS | nuclear localisation signal |
| PAGE | Polyacrylamide gel electrophoresis |
| PBS | Phosphate-Buffered Saline |
| pI | isoelectric point of a protein |
| PTM | post translational modification |
| PTEMF | PIPES, Triton X-100, EGTA, MgCl2, Formaldehyde |
| SDS | Sodium Dodecyl Sulphate |
| SIM | SUMO Interacting Motif |
| SUMO | Small Ubiquitin-like Modifier |
| TBST | Tris-buffered saline with 0.1% Tween-20 |
| TOP2 | Human Topoisomerase II protein |
| TOP2A | human Topoisomerase II alpha protein |
| TOP2A | human Topoisomerase II alpha gene |
| Top2 | S.cerevisiae Topoisomerase II protein |
| Top2a | Xenopus Topoisomerase II alpha protein |
References
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Roca, J. Topoisomerase II: A fitted mechanism for the chromatin landscape. Nucleic Acids Res. 2009, 37, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Higgins, N.P. A human TOP2A core DNA binding X-ray structure reveals topoisomerase subunit dynamics and a potential mechanism for SUMO modulation of decatenation. J. Mol. Biol. 2012, 424, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The structure of DNA-bound human topoisomerase II alpha: Conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012, 424, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.H.; Osheroff, N.; Berger, J.M. Structure of a topoisomerase II-DNA-nucleotide complex reveals a new control mechanism for ATPase activity. Nat. Struct. Mol. Biol. 2012, 19, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Huang, N.L.; Lin, J.H.; Wu, C.C.; Wang, Y.R.; Yu, Y.J.; Gilson, M.K.; Chan, N.L. Structural insights into the gating of DNA passage by the topoisomerase II DNA-gate. Nat. Commun. 2018, 9, 3085. [Google Scholar] [CrossRef] [PubMed]
- Laponogov, I.; Pan, X.S.; Veselkov, D.A.; Skamrova, G.B.; Umrekar, T.R.; Fisher, L.M.; Sanderson, M.R. Trapping of the transport-segment DNA by the ATPase domains of a type II topoisomerase. Nat. Commun. 2018, 9, 2579. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.D.; Drake, F.H.; Tan, K.B.; Per, S.R.; Crooke, S.T.; Mirabelli, C.K. Characterization and immunological identification of cDNA clones encoding two human DNA topoisomerase II isozymes. Proc. Natl. Acad. Sci. USA 1989, 86, 9431–9435. [Google Scholar] [CrossRef]
- Tan, K.B.; Dorman, T.E.; Falls, K.M.; Chung, T.D.; Mirabelli, C.K.; Crooke, S.T.; Mao, J. Topoisomerase II alpha and topoisomerase II beta genes: Characterization and mapping to human chromosomes 17 and 3, respectively. Cancer Res. 1992, 52, 231–234. [Google Scholar]
- Niimi, A.; Suka, N.; Harata, M.; Kikuchi, A.; Mizuno, S. Co-localization of chicken DNA topoisomerase IIalpha, but not beta, with sites of DNA replication and possible involvement of a C-terminal region of alpha through its binding to PCNA. Chromosoma 2001, 110, 102–114. [Google Scholar] [CrossRef]
- Linka, R.M.; Porter, A.C.; Volkov, A.; Mielke, C.; Boege, F.; Christensen, M.O. C-terminal regions of topoisomerase IIalpha and IIbeta determine isoform-specific functioning of the enzymes in vivo. Nucleic Acids Res. 2007, 35, 3810–3822. [Google Scholar] [CrossRef]
- Meczes, E.L.; Gilroy, K.L.; West, K.L.; Austin, C.A. The impact of the human DNA topoisomerase II C-terminal domain on activity. PLoS ONE 2008, 3, e1754. [Google Scholar] [CrossRef]
- Bedez, C.; Lotz, C.; Batisse, C.; Broeck, A.V.; Stote, R.H.; Howard, E.; Pradeau-Aubreton, K.; Ruff, M.; Lamour, V. Post-translational modifications in DNA topoisomerase 2alpha highlight the role of a eukaryote-specific residue in the ATPase domain. Sci. Rep. 2018, 8, 9272. [Google Scholar] [CrossRef]
- Mirski, S.E.; Gerlach, J.H.; Cummings, H.J.; Zirngibl, R.; Greer, P.A.; Cole, S.P. Bipartite nuclear localization signals in the C terminus of human topoisomerase II alpha. Exp. Cell Res. 1997, 237, 452–455. [Google Scholar] [CrossRef]
- Mirski, S.E.; Gerlach, J.H.; Cole, S.P. Sequence determinants of nuclear localization in the alpha and beta isoforms of human topoisomerase II. Exp. Cell Res. 1999, 251, 329–339. [Google Scholar] [CrossRef]
- Dickey, J.S.; Osheroff, N. Impact of the C-terminal domain of topoisomerase IIalpha on the DNA cleavage activity of the human enzyme. Biochemistry 2005, 44, 11546–11554. [Google Scholar] [CrossRef]
- Kozuki, T.; Chikamori, K.; Surleac, M.D.; Micluta, M.A.; Petrescu, A.J.; Norris, E.J.; Elson, P.; Hoeltge, G.A.; Grabowski, D.R.; Porter, A.C.G.; et al. Roles of the C-terminal domains of topoisomerase IIalpha and topoisomerase IIbeta in regulation of the decatenation checkpoint. Nucleic Acids Res. 2017, 45, 5995–6010. [Google Scholar] [CrossRef]
- McClendon, A.K.; Osheroff, N. The geometry of DNA supercoils modulates topoisomerase-mediated DNA cleavage and enzyme response to anticancer drugs. Biochemistry 2006, 45, 3040–3050. [Google Scholar] [CrossRef]
- McClendon, A.K.; Gentry, A.C.; Dickey, J.S.; Brinch, M.; Bendsen, S.; Andersen, A.H.; Osheroff, N. Bimodal recognition of DNA geometry by human topoisomerase II alpha: Preferential relaxation of positively supercoiled DNA requires elements in the C-terminal domain. Biochemistry 2008, 47, 13169–13178. [Google Scholar] [CrossRef]
- Seol, Y.; Gentry, A.C.; Osheroff, N.; Neuman, K.C. Chiral discrimination and writhe-dependent relaxation mechanism of human topoisomerase IIalpha. J. Biol. Chem. 2013, 288, 13695–13703. [Google Scholar] [CrossRef]
- Taagepera, S.; Rao, P.N.; Drake, F.H.; Gorbsky, G.J. DNA topoisomerase II alpha is the major chromosome protein recognized by the mitotic phosphoprotein antibody MPM-2. Proc. Natl. Acad. Sci. USA 1993, 90, 8407–8411. [Google Scholar] [CrossRef]
- Saitoh, N.; Goldberg, I.G.; Wood, E.R.; Earnshaw, W.C. ScII: An abundant chromosome scaffold protein is a member of a family of putative ATPases with an unusual predicted tertiary structure. J. Cell Biol. 1994, 127, 303–318. [Google Scholar] [CrossRef]
- Rattner, J.B.; Hendzel, M.J.; Furbee, C.S.; Muller, M.T.; Bazett-Jones, D.P. Topoisomerase II alpha is associated with the mammalian centromere in a cell cycle- and species-specific manner and is required for proper centromere/kinetochore structure. J. Cell Biol. 1996, 134, 1097–1107. [Google Scholar] [CrossRef] [Green Version]
- Sumner, A.T. The distribution of topoisomerase II on mammalian chromosomes. Chromosome Res. 1996, 4, 5–14. [Google Scholar] [CrossRef]
- Meyer, K.N.; Kjeldsen, E.; Straub, T.; Knudsen, B.R.; Hickson, I.D.; Kikuchi, A.; Kreipe, H.; Boege, F. Cell cycle-coupled relocation of types I and II topoisomerases and modulation of catalytic enzyme activities. J. Cell Biol. 1997, 136, 775–788. [Google Scholar] [CrossRef]
- Christensen, M.O.; Larsen, M.K.; Barthelmes, H.U.; Hock, R.; Andersen, C.L.; Kjeldsen, E.; Knudsen, B.R.; Westergaard, O.; Boege, F.; Mielke, C. Dynamics of human DNA topoisomerases IIalpha and IIbeta in living cells. J. Cell Biol. 2002, 157, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Martinez, L.A.; Gimenez-Abian, J.F.; Azuma, Y.; Guacci, V.; Gimenez-Martin, G.; Lanier, L.M.; Clarke, D.J. PIASgamma is required for faithful chromosome segregation in human cells. PLoS ONE 2006, 1, e53. [Google Scholar] [CrossRef]
- Lane, A.B.; Gimenez-Abian, J.F.; Clarke, D.J. A novel chromatin tether domain controls topoisomerase IIalpha dynamics and mitotic chromosome formation. J. Cell Biol. 2013, 203, 471–486. [Google Scholar] [CrossRef]
- Ryu, H.; Yoshida, M.M.; Sridharan, V.; Kumagai, A.; Dunphy, W.G.; Dasso, M.; Azuma, Y. SUMOylation of the C-terminal domain of DNA topoisomerase IIalpha regulates the centromeric localization of Claspin. Cell Cycle 2015, 14, 2777–2784. [Google Scholar] [CrossRef]
- Yoshida, M.M.; Ting, L.; Gygi, S.P.; Azuma, Y. SUMOylation of DNA topoisomerase IIalpha regulates histone H3 kinase Haspin and H3 phosphorylation in mitosis. J. Cell Biol. 2016, 213, 665–678. [Google Scholar] [CrossRef]
- Kawano, S.; Kato, Y.; Okada, N.; Sano, K.; Tsutsui, K.; Tsutsui, K.M.; Ikeda, S. DNA-binding activity of rat DNA topoisomerase II alpha C-terminal domain contributes to efficient DNA catenation in vitro. J. Biochem. 2016, 159, 363–369. [Google Scholar]
- Wessel, I.; Jensen, P.B.; Falck, J.; Mirski, S.E.; Cole, S.P.; Sehested, M. Loss of amino acids 1490Lys-Ser-Lys1492 in the COOH-terminal region of topoisomerase IIalpha in human small cell lung cancer cells selected for resistance to etoposide results in an extranuclear enzyme localization. Cancer Res. 1997, 57, 4451–4454. [Google Scholar]
- Austin, C.A.; Marsh, K.L. Eukaryotic DNA topoisomerase II beta. Bioessays 1998, 20, 215–226. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef] [Green Version]
- Yavuz, A.S.; Sezerman, O.U. Predicting sumoylation sites using support vector machines based on various sequence features, conformational flexibility and disorder. BMC Genom. 2014, 15 (Suppl. 9), S18. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Lyon, D.; Young, C.; Jensen, L.J.; Vertegaal, A.C.; Nielsen, M.L. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 2017, 24, 325–336. [Google Scholar] [CrossRef]
- Chen, T.; Sun, Y.; Ji, P.; Kopetz, S.; Zhang, W. Topoisomerase IIalpha in chromosome instability and personalized cancer therapy. Oncogene 2015, 34, 4019–4031. [Google Scholar] [CrossRef]
- Azuma, Y.; Arnaoutov, A.; Dasso, M. SUMO-2/3 regulates topoisomerase II in mitosis. J. Cell Biol. 2003, 163, 477–487. [Google Scholar] [CrossRef] [Green Version]
- Azuma, Y.; Arnaoutov, A.; Anan, T.; Dasso, M. PIASy mediates SUMO-2 conjugation of Topoisomerase-II on mitotic chromosomes. EMBO J. 2005, 24, 2172–2182. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.; Furuta, M.; Kirkpatrick, D.; Gygi, S.P.; Azuma, Y. PIASy-dependent SUMOylation regulates DNA topoisomerase IIalpha activity. J. Cell Biol. 2010, 191, 783–794. [Google Scholar] [CrossRef]
- Edgerton, H.; Johansson, M.; Keifenheim, D.; Mukherjee, S.; Chacon, J.M.; Bachant, J.; Gardner, M.K.; Clarke, D.J. A noncatalytic function of the topoisomerase II CTD in Aurora B recruitment to inner centromeres during mitosis. J. Cell Biol. 2016, 213, 651–664. [Google Scholar] [CrossRef] [Green Version]
- Dawlaty, M.M.; Malureanu, L.; Jeganathan, K.B.; Kao, E.; Sustmann, C.; Tahk, S.; Shuai, K.; Grosschedl, R.; van Deursen, J.M. Resolution of sister centromeres requires RanBP2-mediated SUMOylation of topoisomerase IIalpha. Cell 2008, 133, 103–115. [Google Scholar] [CrossRef]
- Carpenter, A.J.; Porter, A.C. Construction, characterization, and complementation of a conditional-lethal DNA topoisomerase IIalpha mutant human cell line. Mol. Biol. Cell 2004, 15, 5700–5711. [Google Scholar] [CrossRef]
- Spence, J.M.; Phua, H.H.; Mills, W.; Carpenter, A.J.; Porter, A.C.; Farr, C.J. Depletion of topoisomerase IIalpha leads to shortening of the metaphase interkinetochore distance and abnormal persistence of PICH-coated anaphase threads. J. Cell Sci. 2007, 120, 3952–3964. [Google Scholar] [CrossRef]
- Farr, C.J.; Antoniou-Kourounioti, M.; Mimmack, M.L.; Volkov, A.; Porter, A.C. The alpha isoform of topoisomerase II is required for hypercompaction of mitotic chromosomes in human cells. Nucleic Acids Res. 2014, 42, 4414–4426. [Google Scholar] [CrossRef]
- Caron, P.R.; Watt, P.; Wang, J.C. The C-terminal domain of Saccharomyces cerevisiae DNA topoisomerase II. Mol. Cell Biol. 1994, 14, 3197–3207. [Google Scholar] [CrossRef]
- Jensen, S.; Andersen, A.H.; Kjeldsen, E.; Biersack, H.; Olsen, E.H.; Andersen, T.B.; Westergaard, O.; Jakobsen, B.K. Analysis of functional domain organization in DNA topoisomerase II from humans and Saccharomyces cerevisiae. Mol. Cell Biol. 1996, 16, 3866–3877. [Google Scholar] [CrossRef]
- Xue, Y.; Zhou, F.; Fu, C.; Xu, Y.; Yao, X. SUMOsp: A web server for sumoylation site prediction. Nucleic Acids Res. 2006, 34, W254–W257. [Google Scholar] [CrossRef]
- Ren, J.; Gao, X.; Jin, C.; Zhu, M.; Wang, X.; Shaw, A.; Wen, L.; Yao, X.; Xue, Y. Systematic study of protein sumoylation: Development of a site-specific predictor of SUMOsp 2.0. Proteomics 2009, 9, 3409–3412. [Google Scholar] [CrossRef]
- Mao, Y.; Desai, S.D.; Liu, L.F. SUMO-1 conjugation to human DNA topoisomerase II isozymes. J. Biol. Chem. 2000, 275, 26066–26073. [Google Scholar] [CrossRef]
- Bachant, J.; Alcasabas, A.; Blat, Y.; Kleckner, N.; Elledge, S.J. The SUMO-1 isopeptidase Smt4 is linked to centromeric cohesion through SUMO-1 modification of DNA topoisomerase II. Mol. Cell 2002, 9, 1169–1182. [Google Scholar] [CrossRef]
- Agostinho, M.; Santos, V.; Ferreira, F.; Costa, R.; Cardoso, J.; Pinheiro, I.; Rino, J.; Jaffray, E.; Hay, R.T.; Ferreira, J. Conjugation of human topoisomerase 2 alpha with small ubiquitin-like modifiers 2/3 in response to topoisomerase inhibitors: Cell cycle stage and chromosome domain specificity. Cancer Res. 2008, 68, 2409–2418. [Google Scholar] [CrossRef] [PubMed]
- Bekes, M.; Prudden, J.; Srikumar, T.; Raught, B.; Boddy, M.N.; Salvesen, G.S. The dynamics and mechanism of SUMO chain deconjugation by SUMO-specific proteases. J. Biol. Chem. 2011, 286, 10238–10247. [Google Scholar] [CrossRef]
- Tatham, M.H.; Rodriguez, M.S.; Xirodimas, D.P.; Hay, R.T. Detection of protein SUMOylation in vivo. Nat. Protoc. 2009, 4, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Hietakangas, V.; Anckar, J.; Blomster, H.A.; Fujimoto, M.; Palvimo, J.J.; Nakai, A.; Sistonen, L. PDSM, a motif for phosphorylation-dependent SUMO modification. Proc. Natl. Acad. Sci. USA 2006, 103, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Mohideen, F.; Capili, A.D.; Bilimoria, P.M.; Yamada, T.; Bonni, A.; Lima, C.D. A molecular basis for phosphorylation-dependent SUMO conjugation by the E2 UBC9. Nat. Struct. Mol. Biol. 2009, 16, 945–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matic, I.; Schimmel, J.; Hendriks, I.A.; van Santen, M.A.; van de Rijke, F.; van Dam, H.; Gnad, F.; Mann, M.; Vertegaal, A.C. Site-specific identification of SUMO-2 targets in cells reveals an inverted SUMOylation motif and a hydrophobic cluster SUMOylation motif. Mol. Cell 2010, 39, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; D’Souza, R.C.; Yang, B.; Verlaan-de Vries, M.; Mann, M.; Vertegaal, A.C. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 2014, 21, 927–936. [Google Scholar] [CrossRef] [Green Version]
- Wells, N.J.; Fry, A.M.; Guano, F.; Norbury, C.; Hickson, I.D. Cell cycle phase-specific phosphorylation of human topoisomerase II alpha. Evidence of a role for protein kinase C. J. Biol. Chem. 1995, 270, 28357–28363. [Google Scholar]
- Kettenbach, A.N.; Schweppe, D.K.; Faherty, B.K.; Pechenick, D.; Pletnev, A.A.; Gerber, S.A. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci. Signal 2011, 4, rs5. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, J.C. Similarity in the catalysis of DNA breakage and rejoining by type IA and IIA DNA topoisomerases. Proc. Natl. Acad. Sci. USA 1999, 96, 881–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, R.; Takashima, R.; Koujin, T.; Shibata, M.; Nozaki, N.; Seto, M.; Mori, H.; Haraguchi, T.; Hiraoka, Y. Mitotic specific phosphorylation of serine-1212 in human DNA topoisomerase IIalpha. Cell Struct. Funct. 2001, 26, 215–226. [Google Scholar] [CrossRef]
- Wells, N.J.; Hickson, I.D. Human topoisomerase II alpha is phosphorylated in a cell-cycle phase-dependent manner by a proline-directed kinase. Eur. J. Biochem. 1995, 231, 491–497. [Google Scholar] [CrossRef]
- Xu, Y.X.; Manley, J.L. The prolyl isomerase Pin1 functions in mitotic chromosome condensation. Mol. Cell 2007, 26, 287–300. [Google Scholar] [CrossRef]
- Vassilev, L.T. Cell cycle synchronization at the G2/M phase border by reversible inhibition of CDK1. Cell Cycle 2006, 5, 2555–2556. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef] [Green Version]
- Tavormina, P.A.; Come, M.G.; Hudson, J.R.; Mo, Y.Y.; Beck, W.T.; Gorbsky, G.J. Rapid exchange of mammalian topoisomerase II alpha at kinetochores and chromosome arms in mitosis. J. Cell Biol. 2002, 158, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Phair, R.D.; Gorski, S.A.; Misteli, T. Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy. Methods Enzymol. 2004, 375, 393–414. [Google Scholar]
- Bulgakova, N.A.; Grigoriev, I.; Yap, A.S.; Akhmanova, A.; Brown, N.H. Dynamic microtubules produce an asymmetric E-cadherin-Bazooka complex to maintain segment boundaries. J. Cell Biol. 2013, 201, 887–901. [Google Scholar] [CrossRef] [Green Version]
- Yanagida, M. Clearing the way for mitosis: Is cohesin a target? Nat. Rev. Mol. Cell Biol. 2009, 10, 489–496. [Google Scholar] [CrossRef]
- Hirota, T.; Gerlich, D.; Koch, B.; Ellenberg, J.; Peters, J.M. Distinct functions of condensin I and II in mitotic chromosome assembly. J. Cell Sci. 2004, 117, 6435–6445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waizenegger, I.C.; Hauf, S.; Meinke, A.; Peters, J.M. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell 2000, 103, 399–410. [Google Scholar] [CrossRef]
- Ruchaud, S.; Carmena, M.; Earnshaw, W.C. Chromosomal passengers: Conducting cell division. Nat. Rev. Mol. Cell Biol. 2007, 8, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, S.G.; Barber, C.M.; Allis, C.D.; Sullivan, K.F. Differential regulation of CENP-A and histone H3 phosphorylation in G2/M. J. Cell Sci. 2001, 114, 653–661. [Google Scholar] [PubMed]
- Monier, K.; Mouradian, S.; Sullivan, K.F. DNA methylation promotes Aurora-B-driven phosphorylation of histone H3 in chromosomal subdomains. J. Cell Sci. 2007, 120, 101–114. [Google Scholar] [CrossRef]
- Polioudaki, H.; Markaki, Y.; Kourmouli, N.; Dialynas, G.; Theodoropoulos, P.A.; Singh, P.B.; Georgatos, S.D. Mitotic phosphorylation of histone H3 at threonine 3. FEBS Lett. 2004, 560, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Sultan, S.; Taylor, S.S.; Higgins, J.M. The kinase haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev. 2005, 19, 472–488. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Yong-Gonzalez, V.; Kikuchi, Y.; Strunnikov, A. SIZ1/SIZ2 control of chromosome transmission fidelity is mediated by the sumoylation of topoisomerase II. Genetics 2006, 172, 783–794. [Google Scholar] [CrossRef]
- Tammsalu, T.; Matic, I.; Jaffray, E.G.; Ibrahim, A.F.M.; Tatham, M.H.; Hay, R.T. Proteome-wide identification of SUMO2 modification sites. Sci. Signal 2014, 7, rs2. [Google Scholar] [CrossRef]
- Impens, F.; Radoshevich, L.; Cossart, P.; Ribet, D. Mapping of SUMO sites and analysis of SUMOylation changes induced by external stimuli. Proc. Natl. Acad. Sci. USA 2014, 111, 12432–12437. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, I.A.; D’Souza, R.C.; Chang, J.G.; Mann, M.; Vertegaal, A.C. System-wide identification of wild-type SUMO-2 conjugation sites. Nat. Commun. 2015, 6, 7289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Z.; Chang, J.G.; Hendriks, I.A.; Sigurethsson, J.O.; Olsen, J.V.; Vertegaal, A.C. System-wide Analysis of SUMOylation Dynamics in Response to Replication Stress Reveals Novel Small Ubiquitin-like Modified Target Proteins and Acceptor Lysines Relevant for Genome Stability. Mol. Cell Proteom. 2015, 14, 1419–1434. [Google Scholar] [CrossRef] [PubMed]
- Lumpkin, R.J.; Gu, H.; Zhu, Y.; Leonard, M.; Ahmad, A.S.; Clauser, K.R.; Meyer, J.G.; Bennett, E.J.; Komives, E.A. Site-specific identification and quantitation of endogenous SUMO modifications under native conditions. Nat. Commun. 2017, 8, 1171. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; Lyon, D.; Su, D.; Skotte, N.H.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Site-specific characterization of endogenous SUMOylation across species and organs. Nat. Commun. 2018, 9, 2456. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.; Bachant, J. SUMO modification of DNA topoisomerase II: Trying to get a CENse of it all. DNA Repair (Amst.) 2009, 8, 557–568. [Google Scholar] [CrossRef]
- Clarke, D.J.; Azuma, Y. Non-Catalytic Roles of the Topoisomerase IIalpha C-Terminal Domain. Int. J. Mol. Sci. 2017, 18, 2438. [Google Scholar] [CrossRef]
- Tatham, M.H.; Jaffray, E.; Vaughan, O.A.; Desterro, J.M.; Botting, C.H.; Naismith, J.H.; Hay, R.T. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 2001, 276, 35368–35374. [Google Scholar] [CrossRef]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Polo, S.; Miller, K.M.; Jackson, S.P. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 2009, 462, 935–939. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Expression Construct | Residues Deleted | Complementation (Growth in Dox) | TOP2A Localisation * | |
|---|---|---|---|---|
| Interphase | M Phase | |||
| FT (Flag:TOP2A) | none | Y | Nuc | Arm + cen |
| FTΔ2 | 1173–1446 | N | N + C | diffuse |
| FTΔ3 | 1321–1446 | Y | Nuc | Arm + cen |
| FTΔ5 | 1212–1446 | Y | Nuc | diffuse |
| Cell Line | Fast t1/2 (s) * | Slow t1/2 (s) * | Immobile Fraction | Curve Fitting Compared to GFP:TOP2A | |
|---|---|---|---|---|---|
| Goodness of Fit (R Square) | Significance (p Value) | ||||
| GFP:TOP2A | 5.15 ± 1.13 | 33.49 ± 2.86 | 6.71 × 10−11 | 0.997 | NS (>0.9999) |
| GT-K662R | 8.12 ± 2.59 | 52.98 ± 11.76 | 1.14 × 10−16 | 0.892 | <0.0001 |
| GT:SUMO2 | 2.59 ± 1.24 | 12.82 ± 1.20 | 1.33 × 10−16 | 0.530 | <0.0001 |
| G:SUMO2:T | 2.53 ± 1.05 | 23.87 ± 1.24 | 1.25 × 10−16 | 0.947 | <0.0001 |
| GT-K1240R | 2.87 ± 2.30 | 31.47 ± 2.64 | 1.72 × 10−16 | 0.945 | NS (0.4219) |
| GT-S1247A | 5.16 ± 1.91 | 34.03 ± 3.00 | 1.22 × 10−16 | 0.974 | NS (0.8093) |
| GT-S1213A | 16.17 ± 4.11 | 85.14 ± 43.43 | 0.066 | 0.186 | <0.0001 |
| Cell Line | Fast t1/2 (s) * | Slow t1/2 (s) * | Immobile Fraction | Curve Fitting Compared to GFP:TOP2A | |
|---|---|---|---|---|---|
| Goodness of Fit (R Square) | Significance (p Value) | ||||
| GFP:TOP2A | 9.4 ± 2.1 | 61.15 ± 14.96 | 0.015 | 0.968 | NS (0.9632) |
| GT-S1213A | 10.47 ± 2.94 | 71.87 ± 15.09 | 0.035 | 0.653 | <0.0001 |
| GT-S1213D | 9.31 ± 1.69 | 45.45 ± 9.73 | 0.055 | 0.976 | NS (0.7924) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antoniou-Kourounioti, M.; Mimmack, M.L.; Porter, A.C.G.; Farr, C.J. The Impact of the C-Terminal Region on the Interaction of Topoisomerase II Alpha with Mitotic Chromatin. Int. J. Mol. Sci. 2019, 20, 1238. https://doi.org/10.3390/ijms20051238
Antoniou-Kourounioti M, Mimmack ML, Porter ACG, Farr CJ. The Impact of the C-Terminal Region on the Interaction of Topoisomerase II Alpha with Mitotic Chromatin. International Journal of Molecular Sciences. 2019; 20(5):1238. https://doi.org/10.3390/ijms20051238
Chicago/Turabian StyleAntoniou-Kourounioti, Melissa, Michael L. Mimmack, Andrew C.G. Porter, and Christine J. Farr. 2019. "The Impact of the C-Terminal Region on the Interaction of Topoisomerase II Alpha with Mitotic Chromatin" International Journal of Molecular Sciences 20, no. 5: 1238. https://doi.org/10.3390/ijms20051238
APA StyleAntoniou-Kourounioti, M., Mimmack, M. L., Porter, A. C. G., & Farr, C. J. (2019). The Impact of the C-Terminal Region on the Interaction of Topoisomerase II Alpha with Mitotic Chromatin. International Journal of Molecular Sciences, 20(5), 1238. https://doi.org/10.3390/ijms20051238