ROCK2 Regulates Monocyte Migration and Cell to Cell Adhesion in Vascular Endothelial Cells


 ,
,
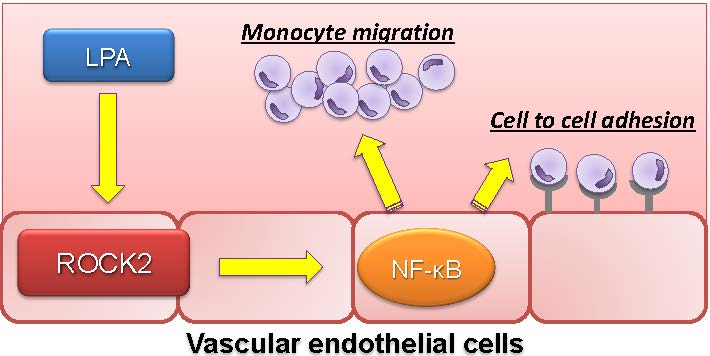
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
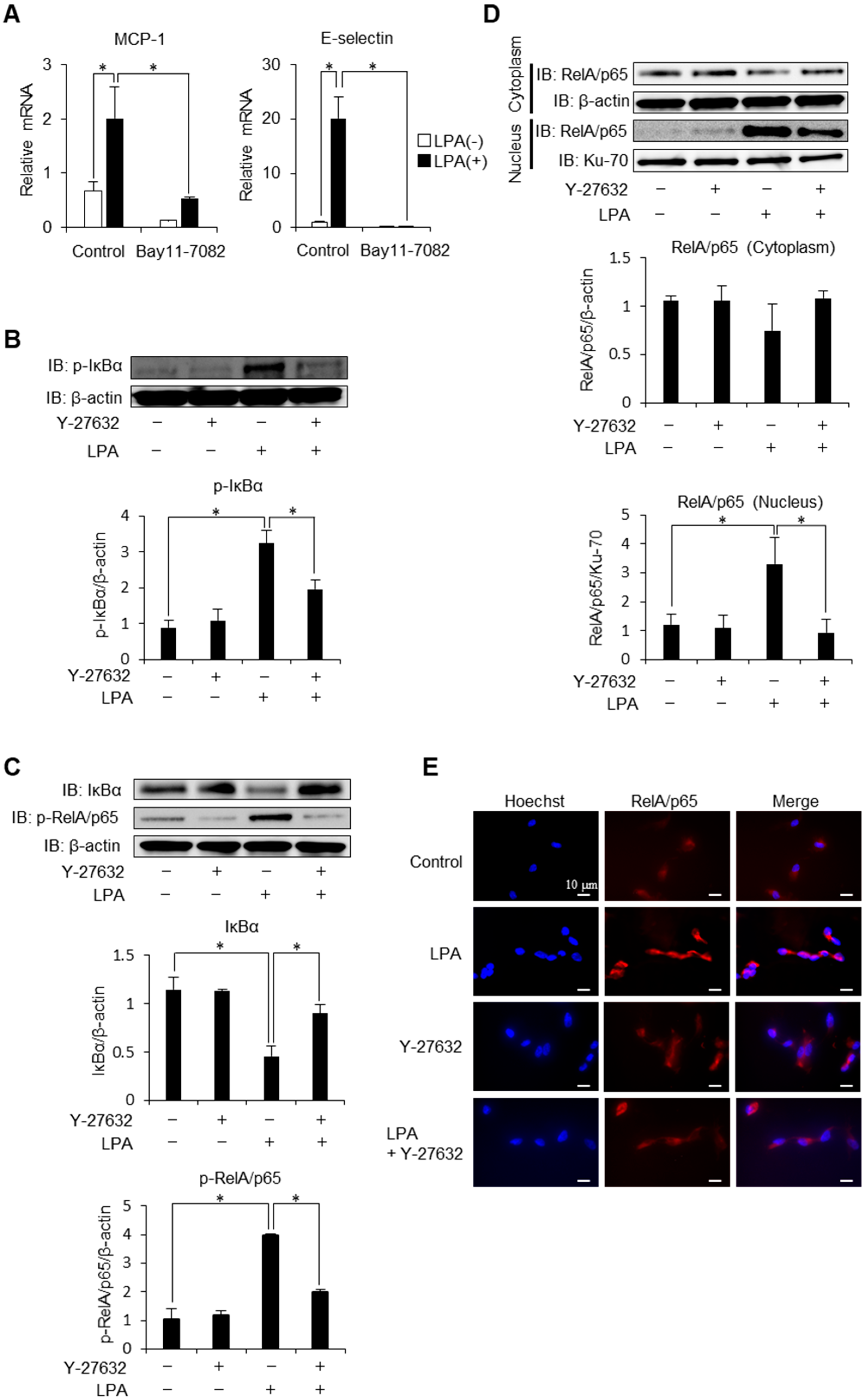
2.1. Regulation of Inflammatory Cytokines, Receptors and Adhesion Molecules via ROCK
2.2. Phosphorylation of IκBα is Mediated via ROCK Signaling
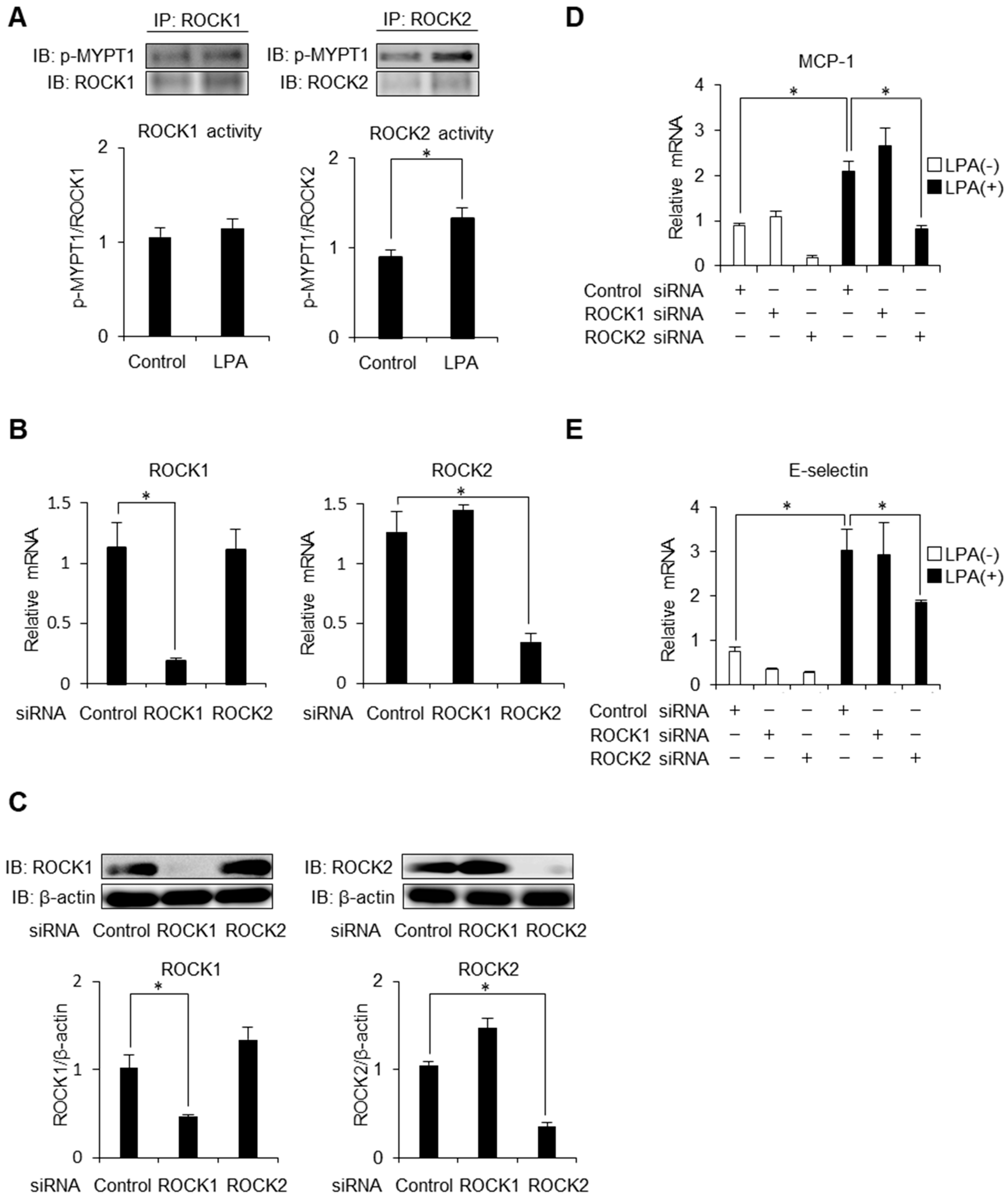
2.3. ROCK2 is Required for LPA-Induced MCP-1 and E-Selectin Expression in HAECs
2.4. Endothelial ROCK2 Regulates Recruitment of Monocytic Cells
2.5. ROCK2 Controls Cell to Cell Adhesion in HAECs
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. PCR Array
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. RNA Isolation and Quantitative Real-Time PCR
4.6. Western Blot Analysis
4.7. Transfection and Reporter Gene Assay
4.8. Immunocytochemistry
4.9. RNA Interference
4.10. Immunoprecipitation
4.11. Chemotaxis Assay
4.12. Adhesion Assay
4.13. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ROCK | Rho-kinase |
| LPA | lysophosphatidic acid |
| CAMs | chemokines and cell adhesion molecules |
| MCP-1 | monocyte chemoattractant protein-1 |
| HAECs | human aortic endothelial cells |
| VCAM-1 | vascular cell adhesion molecule-1 |
| ICAM-1 | intracellular adhesion molecule-1 |
| MYPT1 | myosin phosphatase target subunit 1 |
| siRNA | small interfering RNA |
| IKK | IκB kinase |
| VSMC | vascular smooth muscle cell |
References
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Rajagopalan, L.E. Rho kinase-2 activation in human endothelial cells driveslysophosphatidic acid-mediated expression of cell adhesion molecules via NF-kappaB p65. J. Biol. Chem. 2010, 285, 12536–12542. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tong, J.; He, D.; Pendyala, S.; Evgeny, B.; Chun, J.; Sperling, A.I.; Natarajan, V. Role of lysophosphatidic acid receptor LPA2 in the development of allergic airway inflammation in a murine model of asthma. Respir. Res. 2009, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Miyauchi, K.; Ohkawa, R.; Nakamura, K.; Kishimoto, T.; Miyazaki, T.; Nishino, A.; Nakajima, N.; Yaginuma, K.; Tamura, H.; et al. Increased circulating plasma lysophosphatidic acid in patients with acute coronary syndrome. Clin. Chim. Acta 2012, 413, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Oh, Y.S.; Choi, J.W.; Jung, J.Y.; Jun, H.S. Blocking lysophosphatidic acid receptor 1 signaling inhibits diabetic nephropathy in db/db mice. Kidney Int. 2017, 91, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Siess, W.; Zangl, K.J.; Essler, M.; Bauer, M.; Brandl, R.; Corrinth, C.; Bittman, R.; Tigyi, G.; Aepfelbacher, M. Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc. Natl. Acad. Sci. USA 1999, 96, 6931–6936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bot, M.; Bot, I.; Lopez-Vales, R.; van de Lest, C.H.; Saulnier-Blache, J.S.; Helms, J.B.; David, S.; van Berkel, T.J.; Biessen, E.A. Atherosclerotic lesion progression changes lysophosphatidic acid homeostasis to favor its accumulation. Am. J. Pathol. 2010, 176, 3073–3084. [Google Scholar] [CrossRef]
- Schober, A.; Siess, W. Lysophosphatidic acid in atherosclerotic diseases. Br. J. Pharmacol. 2012, 167, 465–482. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Subramanian, P.; Sevilmis, G.; Globke, B.; Soehnlein, O.; Karshovska, E.; Megens, R.; Heyll, K.; Chun, J.; Saulnier-Blache, J.S.; et al. Lipoprotein-derived lysophosphatidic acid promotes atherosclerosis by releasing CXCL1 from the endothelium. Cell Metab. 2011, 13, 592–600. [Google Scholar] [CrossRef]
- Chou, C.H.; Lai, S.L.; Ho, C.M.; Lin, W.H.; Chen, C.N.; Lee, P.H.; Peng, F.C.; Kuo, S.H.; Wu, S.Y.; Lai, H.S. Lysophosphatidic acid alters the expression profiles of angiogenic factors, cytokines, and chemokines in mouse liver sinusoidal endothelial cells. PLoS ONE 2015, 10, e0122060. [Google Scholar] [CrossRef]
- Kritikou, E.; van Puijvelde, G.H.; van der Heijden, T.; van Santbrink, P.J.; Swart, M.; Schaftenaar, F.H.; Kröner, M.J.; Kuiper, J.; Bot, I. Inhibition of lysophosphatidic acid receptors 1 and 3 attenuates atherosclerosis development in LDL-receptor deficient mice. Sci. Rep. 2016, 6, 37585. [Google Scholar] [CrossRef] [Green Version]
- Amano, M.; Chihara, K.; Kimura, K.; Fukata, Y.; Nakamura, N.; Matsuura, Y.; Kaibuchi, K. Formation of Actin Stress Fibers and Focal Adhesions Enhanced by Rho-Kinase. Science 1997, 275, 1308–1311. [Google Scholar] [CrossRef]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K.; et al. Regulation of Myosin Phosphatase by Rho and Rho-Associated Kinase (Rho-Kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Kamiyama, M.; Utsunomiya, K.; Taniguchi, K.; Yokota, T.; Kurata, H.; Tajima, N.; Kondo, K. Contribution of Rho A and Rho Kinase to Platelet-Derived Growth Factor–BB-Induced Proliferation of Vascular Smooth Muscle Cells. J. Atheroscler. Thromb. 2003, 10, 117–123. [Google Scholar] [CrossRef]
- Shimokawa, H.; Takeshita, A. Rho-kinase is an important therapeutic target in cardiovascular medicine. Arter. Thromb. Vasc. Biol. 2005, 25, 1767–1775. [Google Scholar] [CrossRef]
- Kawanami, D.; Matoba, K.; Kanazawa, Y.; Ishizawa, S.; Yokota, T.; Utsunomiya, K. Thrombin induces MCP-1 expression through Rho-kinase and subsequent p38MAPK/NF-kappaB signaling pathway activation in vascular endothelial cells. Biochem. Biophys. Res. Commun. 2011, 411, 798–803. [Google Scholar] [CrossRef]
- Nakagawa, O.; Fujisawa, K.; Ishizaki, T.; Saito, Y.; Nakao, K.; Narumiya, S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996, 392, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Zingarelli, B.; Harris, V.; Tempel, G.E.; Halushka, P.V.; Cook, J.A. Lysophosphatidic Acid Inhibits Bacterial Endotoxin-Induced Pro-Inflammatory Response: Potential Anti Inflammatory Signaling Pathways. Mol. Med. 2008, 14, 422–428. [Google Scholar] [CrossRef]
- Plastira, I.; Bernhart, E.; Goeritzer, M.; Reicher, H.; Kumble, V.B.; Kogelnik, N.; Wintersperger, A.; Hammer, A.; Schlager, S.; Jandl, K.; et al. 1-Oleyl-lysophosphatidic acid (LPA) promotes polarization of BV-2 and primary murine microglia towards an M1-like phenotype. J. Neuroinflammation 2016, 13, 205. [Google Scholar] [CrossRef]
- Gosling, J.; Slaymaker, S.; Gu, L.; Tseng, S.; Zlot, C.H.; Young, S.G.; Rollins, B.J.; Charo, I.F. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J. Clin. Investig. 1999, 103, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.J.; Ballantyne, C.M.; Sharrett, A.R.; Smith, L.C.; Davis, C.E.; Gotto, A.M., Jr.; Boerwinkle, E. Circulating Adhesion Molecules VCAM-1, ICAM-1, and E-selectin in Carotid Atherosclerosis and Incident Coronary Heart Disease Cases: The Atherosclerosis Risk In Communities (ARIC) study. Circulation 1997, 96, 4219–4225. [Google Scholar] [CrossRef]
- Mulero, M.C.; Huang, D.B.; Nguyen, H.T.; Wang, V.Y.; Li, Y.; Biswas, T.; Ghosh, G. DNA-binding affinity and transcriptional activity of the RelA homodimer of nuclear factor κB are not correlated. J. Biol. Chem. 2017, 292, 18821–18830. [Google Scholar] [CrossRef]
- Ueda, A.; Ishigatsubo, Y.; Okubo, T.; Yoshimura, T. Transcriptional regulation of the human monocyte chemoattractant protein-1 gene. Cooperation of two NF-κB sites and NF-κB/Rel subunit specificity. J. Biol. Chem. 1997, 272, 31092–31099. [Google Scholar] [CrossRef]
- Shimada, H.; Rajagopalan, L.E. Rho-kinase mediates lysophosphatidic acid-induced IL-8 and MCP-1 production via p38 and JNK pathways in human endothelial cells. FEBS Lett. 2010, 584, 2827–2832. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.Y.; Zuraw, B.L.; Ye, R.D.; Pan, Z.K. A Rho exchange factor mediates fMet-Leu-Phe-induced NF-kappaB activation in human peripheral blood monocytes. J. Biol. Chem. 2004, 279, 7208–7212. [Google Scholar] [CrossRef]
- Fazal, F.; Bijli, K.M.; Minhajuddin, M.; Rein, T.; Finkelstein, J.N.; Rahman, A. Essential role of cofilin-1 in regulating thrombin-induced RelA/p65 nuclear translocation and intercellular adhesion molecule 1 (ICAM-1) expression in endothelial cells. J. Biol. Chem. 2009, 284, 21047–21056. [Google Scholar] [CrossRef]
- Meyer-Schwesinger, C.; Dehde, S.; von Ruffer, C.; Gatzemeier, S.; Klug, P.; Wenzel, U.O.; Stahl, R.A.; Thaiss, F.; Meyer, T.N. Rho kinase inhibition attenuates LPS-induced renal failure in mice in part by attenuation of NF-kappaB p65 signaling. Am. J. Physiol. Ren. Physiol. 2009, 296, F1088–F1099. [Google Scholar] [CrossRef]
- Brown, K.; Gerstberger, S.; Carlson, L.; Franzoso, G.; Siebenlist, U. Control of IκBα Proteolysis by Site-Specific, Signal-induced Phosphorylation. Science 1995, 267, 1485–1488. [Google Scholar] [CrossRef]
- Traenckner, E.B.; Pahl, H.L.; Henkel, T.; Schmidt, K.N.; Wilk, S.; Baeuerle, P.A. Phosphorylation of human IκB-α on serines 32 and 36 controls IκB-α proteolysis and NF-κB activation in response to diverse stimuli. EMBO J. 1995, 14, 2876–2883. [Google Scholar] [CrossRef]
- Schweitzer, K.; Bozko, P.M.; Dubiel, W.; Naumann, M. CSN controls NF-κB by deubiquitinylation of IκBα. EMBO J. 2007, 26, 1532–1541. [Google Scholar] [CrossRef] [Green Version]
- Panday, A.; Inda, M.E.; Bagam, P.; Sahoo, M.K.; Osorio, D.; Batra, S. Transcription Factor NF-kappaB: An Update on Intervention Strategies. Arch. Immunol. Exp. 2016, 64, 463–483. [Google Scholar] [CrossRef]
- Anwar, K.N.; Fazal, F.; Malik, A.B.; Rahman, A. RhoA/Rho-Associated Kinase Pathway Selectively Regulates Thrombin-Induced Intercellular Adhesion Molecule-1 Expression in Endothelial Cells via Activation of IκB Kinase and Phosphorylation of RelA/p65. J. Immunol. 2004, 173, 6965–6972. [Google Scholar] [CrossRef]
- Matoba, K.; Kawanami, D.; Tsukamoto, M.; Kinoshita, J.; Ito, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Murai, N.; Matsufuji, S.; Utsunomiya, K. Rho-kinase regulation of TNF-α-induced nuclear translocation of NF-κB RelA/p65 and M-CSF expression via p38 MAPK in mesangial cells. Am. J. Physiol. Ren. Physiol. 2014, 307, F571–F580. [Google Scholar] [CrossRef] [Green Version]
- Antoniellis Silveila, A.A.; Dominical, V.M.; Morelli Vital, D.; Alves Ferreira, W., Jr.; Trindade Maranhao Costa, F.; Werneck, C.C.; Ferreira Costa, F.; Conran, N. Attenuation of TNF-induced neutrophil adhesion by simvastatin is associated with the inhibition of Rho-GTPase activity, p50 activity and morphological changes. Int. Immunopharamacol. 2018, 58, 160–165. [Google Scholar] [CrossRef]
- Pelosi, M.; Marampon, F.; Zani, B.M.; Prudente, S.; Perlas, E.; Caputo, V.; Cianetti, L.; Berno, V.; Narumiya, S.; Kang, S.W.; et al. ROCK2 and its alternatively spliced isoform ROCK2m positively control the maturation of the myogenic program. Mol. Cell. Biol. 2007, 17, 6163–6176. [Google Scholar] [CrossRef]
- Beckers, C.M.; Knezevic, N.; Valent, E.T.; Tauseef, M.; Krishnan, R.; Rajendran, K.; Hardin, C.C.; Aman, J.; van Bezu, J.; Sweetnam, P.; et al. ROCK2 primes the endothelium for vascular hyperpermeability responses by raising baseline junctional tension. Vasc. Pharmacol. 2015, 70, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Sawada, N.; Liao, J.K. Rho/Rho-associated coiled-coil forming kinase pathway as therapeutic targets for statins in atherosclerosis. Antioxid. Redox Signal. 2014, 20, 1251–1267. [Google Scholar] [CrossRef]
- Shimizu, T.; Fukumoto, Y.; Tanaka, S.; Satoh, K.; Ikeda, S.; Shimokawa, H. Crucial role of ROCK2 in vascular smooth muscle cells for hypoxia-induced pulmonary hypertension in mice. Arter. Thromb. Vasc. Biol. 2013, 33, 2780–2791. [Google Scholar] [CrossRef]
- Satoh, S.; Takayasu, M.; Kawasaki, K.; Ikegaki, I.; Hitomi, A.; Yano, K.; Shibuya, M.; Asano, T. Antivasospastic Effects of Hydroxyfasudil, a Rho-Kinase Inhibitor, After Subarachnoid Hemorrhage. J. Pharm. Sci. 2012, 118, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; LoGrasso, P.V.; Defert, O.; Li, R. Rho Kinase (ROCK) Inhibitors and Their Therapeutic Potential. J. Med. Chem. 2016, 59, 2269–2300. [Google Scholar] [CrossRef] [PubMed]
- Garnock-Jones, K.P. Ripasudil: First global approval. Drugs 2014, 74, 2211–2215. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Zheng, Y.; von Bornstadt, D.; Wei, Y.; Balcioglu, A.; Daneshmand, A.; Yalcin, N.; Yu, E.; Herisson, F.; Atalay, Y.B.; et al. Selective ROCK2 Inhibition In Focal Cerebral Ischemia. Ann. Clin. Transl. Neurol. 2014, 1, 2–14. [Google Scholar] [PubMed]
- Zanin-Zhorov, A.; Weiss, J.M.; Nyuydzefe, M.S.; Chen, W.; Scher, J.U.; Mo, R.; Depoil, D.; Rao, N.; Liu, B.; Wei, J.; et al. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, 16814–16819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, R.; Paz, K.; Du, J.; Reichenbach, D.K.; Taylor, P.A.; Panoskaltsis-Mortari, A.; Vulic, A.; Luznik, L.; MacDonald, K.K.; Hill, G.R.; et al. Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism. Blood 2016, 127, 2144–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanin-Zhorov, A.; Weiss, J.M.; Trzeciak, A.; Chen, W.; Zhang, J.; Nyuydzefe, M.S.; Arencibia, C.; Polimera, S.; Schueller, O.; Fuentes-Duculan, J.; et al. Cutting edge: Selective oral ROCK2 inhibitor reduces clinical scores in patients with psoriasis vulgaris and normalizes skin pathology via concurrent regulation of IL-17 and IL-10. J. Immunol. 2017, 198, 3809–3814. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, P.; Karshovska, E.; Reinhard, P.; Megens, R.T.; Zhou, Z.; Akhtar, S.; Schumann, U.; Li, X.; van Zandvoort, M.; Ludin, C.; et al. Lysophosphatidic acid receptors LPA1 and LPA3 promote CXCL12-mediated smooth muscle progenitor cell recruitment in neointima formation. Circ. Res. 2010, 107, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Sardella, A.; Chun, J.; Poubelle, P.E.; Fernandes, M.J.; Bourgoin, S.G. TNF-alpha promotes LPA1- and LPA3-mediated recruitment of leukocytes in vivo through CXCR2 ligand chemokines. J. Lipid Res. 2011, 52, 1307–1318. [Google Scholar] [CrossRef]
- Miyata, K.; Shimokawa, H.; Kandabashi, T.; Higo, T.; Morishige, K.; Eto, Y.; Egashira, K.; Kaibuchi, K.; Takeshita, A. Rho-kinase is involved in macrophage-mediated formation of coronary vascular lesions in pigs in vivo. Arterioscler. Hromb. Vasc. Biol. 2000, 20, 2351–2358. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeda, Y.; Matoba, K.; Kawanami, D.; Nagai, Y.; Akamine, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. ROCK2 Regulates Monocyte Migration and Cell to Cell Adhesion in Vascular Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 1331. https://doi.org/10.3390/ijms20061331
Takeda Y, Matoba K, Kawanami D, Nagai Y, Akamine T, Ishizawa S, Kanazawa Y, Yokota T, Utsunomiya K. ROCK2 Regulates Monocyte Migration and Cell to Cell Adhesion in Vascular Endothelial Cells. International Journal of Molecular Sciences. 2019; 20(6):1331. https://doi.org/10.3390/ijms20061331
Chicago/Turabian StyleTakeda, Yusuke, Keiichiro Matoba, Daiji Kawanami, Yosuke Nagai, Tomoyo Akamine, Sho Ishizawa, Yasushi Kanazawa, Tamotsu Yokota, and Kazunori Utsunomiya. 2019. "ROCK2 Regulates Monocyte Migration and Cell to Cell Adhesion in Vascular Endothelial Cells" International Journal of Molecular Sciences 20, no. 6: 1331. https://doi.org/10.3390/ijms20061331
APA StyleTakeda, Y., Matoba, K., Kawanami, D., Nagai, Y., Akamine, T., Ishizawa, S., Kanazawa, Y., Yokota, T., & Utsunomiya, K. (2019). ROCK2 Regulates Monocyte Migration and Cell to Cell Adhesion in Vascular Endothelial Cells. International Journal of Molecular Sciences, 20(6), 1331. https://doi.org/10.3390/ijms20061331