Ion Channels Involved in Substance P-Mediated Nociception and Antinociception



Abstract
:1. Background
2. SP-Mediated Signaling
3. SP and Pain
4. SP-Mediated Anti-Nociception
5. SP-Mediated Anti-Nociception in Muscle
6. Ion Channels Involved in SP Signaling
7. Ion Channels Involved in SP-Mediated Nociception
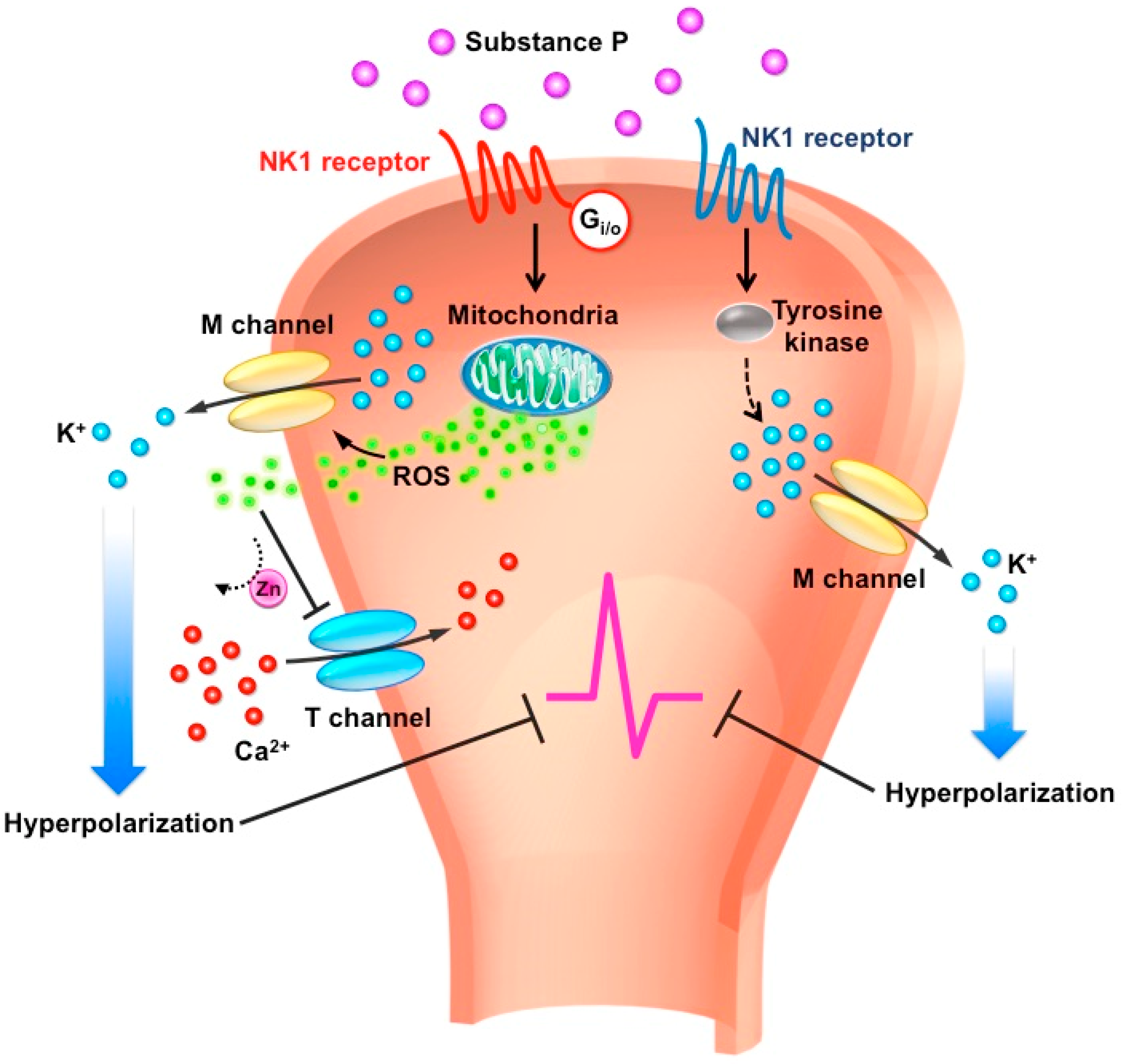
8. Ion Channels Involved in SP-Mediated Anti-Nociception
9. M-type Potassium Channels
10. T-type Calcium Channels
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Euler, U.S.; Gaddum, J.H. An unidentified depressor substance in certain tissue extracts. J. Physiol. 1931, 72, 74–87. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.M.; Leeman, S.E. Isolation of a sialogogic peptide from bovine hypothalamic tissue and its characterization as substance P. J. Biol. Chem. 1970, 245, 4784–4790. [Google Scholar] [PubMed]
- Chang, M.M.; Leeman, S.E.; Niall, H.D. Amino-acid sequence of substance P. Nat. New Biol. 1971, 232, 86–87. [Google Scholar] [CrossRef] [PubMed]
- Krause, J.E.; Chirgwin, J.M.; Carter, M.S.; Xu, Z.S.; Hershey, A.D. Three rat preprotachykinin mRNAs encode the neuropeptides substance P and neurokinin A. Proc. Natl. Acad. Sci. USA 1987, 84, 881–885. [Google Scholar] [CrossRef]
- Jessell, T.M. Substance P in Nociceptive Sensory Neurons. In Substance P in the Nervous System; Porter, R., O’Connor, M., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 1982. [Google Scholar]
- Rameshwar, P.; Gascon, P.; Ganea, D. Immunoregulatory effects of neuropeptides. Stimulation of interleukin-2 production by substance p. J. Neuroimmunol. 1992, 37, 65–74. [Google Scholar] [CrossRef]
- Potter, G.D.; Guzman, F.; Lim, R.K. Visceral pain evoked by intra-arterial injection of substance P. Nature 1962, 193, 983–984. [Google Scholar] [CrossRef] [PubMed]
- Jessell, T.M. Neurotransmitters and CNS disease. Pain. Lancet 1982, 2, 1084–1088. [Google Scholar] [CrossRef]
- Iversen, L. Substance P equals pain substance? Nature 1998, 392, 334–335. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.M.; Getto, C.J.; Neldner, K.; Reeve, E.B.; Krivoy, W.A.; Zimmermann, E. Substance P and analgesia. Nature 1976, 262, 784–785. [Google Scholar] [CrossRef]
- Hall, M.E.; Stewart, J.M. Substance P and antinociception. Peptides 1983, 4, 31–35. [Google Scholar] [CrossRef]
- Macdonald, S.G.; Dumas, J.J.; Boyd, N.D. Chemical cross-linking of the substance P (NK-1) receptor to the alpha subunits of the G proteins Gq and G11. Biochemistry 1996, 35, 2909–2916. [Google Scholar] [CrossRef]
- Meza, U.; Thapliyal, A.; Bannister, R.A.; Adams, B.A. Neurokinin 1 receptors trigger overlapping stimulation and inhibition of CaV2.3 (R-type) calcium channels. Mol. Pharmacol. 2007, 71, 284–293. [Google Scholar] [CrossRef]
- Meshki, J.; Douglas, S.D.; Lai, J.P.; Schwartz, L.; Kilpatrick, L.E.; Tuluc, F. Neurokinin 1 receptor mediates membrane blebbing in HEK293 cells through a Rho/Rho-associated coiled-coil kinase-dependent mechanism. J. Biol. Chem. 2009, 284, 9280–9289. [Google Scholar] [CrossRef]
- Roush, E.D.; Kwatra, M.M. Human substance P receptor expressed in Chinese hamster ovary cells directly activates G(alpha q/11), G(alpha s), G(alpha o). FEBS Lett. 1998, 428, 291–294. [Google Scholar] [CrossRef]
- Khawaja, A.M.; Rogers, D.F. Tachykinins: Receptor to effector. Int. J. Biochem. Cell Biol. 1996, 28, 721–738. [Google Scholar] [CrossRef]
- Fong, T.M.; Anderson, S.A.; Yu, H.; Huang, R.R.; Strader, C.D. Differential activation of intracellular effector by two isoforms of human neurokinin-1 receptor. Mol. Pharmacol. 1992, 41, 24–30. [Google Scholar] [PubMed]
- Baker, S.J.; Morris, J.L.; Gibbins, I.L. Cloning of a C-terminally truncated NK-1 receptor from guinea-pig nervous system. Brain Res. Mol. Brain Res. 2003, 111, 136–147. [Google Scholar] [CrossRef]
- DeFea, K.A.; Vaughn, Z.D.; O’Bryan, E.M.; Nishijima, D.; Dery, O.; Bunnett, N.W. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta -arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. USA 2000, 97, 11086–11091. [Google Scholar] [CrossRef]
- Lai, J.P.; Lai, S.; Tuluc, F.; Tansky, M.F.; Kilpatrick, L.E.; Leeman, S.E.; Douglas, S.D. Differences in the length of the carboxyl terminus mediate functional properties of neurokinin-1 receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 12605–12610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caberlotto, L.; Hurd, Y.L.; Murdock, P.; Wahlin, J.P.; Melotto, S.; Corsi, M.; Carletti, R. Neurokinin 1 receptor and relative abundance of the short and long isoforms in the human brain. Eur. J. Neurosci. 2003, 17, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Harstall, C.; Ospina, M. How prevalent is chronic pain. Pain Clin. Updates 2003, 11, 1–4. [Google Scholar]
- Tracey, D.J.; Walker, J.S. Pain Due to Nerve Damage—Are Inflammatory Mediators Involved. Inflamm. Res. 1995, 44, 407–411. [Google Scholar] [CrossRef]
- Moalem, G.; Tracey, D.J. Immune and inflammatory mechanisms in neuropathic pain. Brain Res. Rev. 2006, 51, 240–264. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.J. Can we distinguish between inflammatory and neuropathic pain? Pain Res. Manag. 2006, 11, 11A–15A. [Google Scholar] [CrossRef]
- De Koninck, Y.; Henry, J.L. Substance P-mediated slow excitatory postsynaptic potential elicited in dorsal horn neurons in vivo by noxious stimulation. Proc. Natl. Acad. Sci. USA 1991, 88, 11344–11348. [Google Scholar] [CrossRef] [PubMed]
- Holzer, P. Neurogenic vasodilatation and plasma leakage in the skin. Gen. Pharmacol. 1998, 30, 5–11. [Google Scholar] [CrossRef]
- Otsuka, M.; Yoshioka, K. Neurotransmitter functions of mammalian tachykinins. Physiol. Rev. 1993, 73, 229–308. [Google Scholar] [CrossRef] [PubMed]
- Russell, I.J.; Orr, M.D.; Littman, B.; Vipraio, G.A.; Alboukrek, D.; Michalek, J.E.; Lopez, Y.; MacKillip, F. Elevated cerebrospinal fluid levels of substance P in patients with the fibromyalgia syndrome. Arthritis Rheumatol. 1994, 37, 1593–1601. [Google Scholar] [CrossRef]
- Vaeroy, H.; Helle, R.; Forre, O.; Kass, E.; Terenius, L. Elevated CSF levels of substance P and high incidence of Raynaud phenomenon in patients with fibromyalgia: New features for diagnosis. Pain 1988, 32, 21–26. [Google Scholar] [CrossRef]
- Mantyh, P.W.; Rogers, S.D.; Honore, P.; Allen, B.J.; Ghilardi, J.R.; Li, J.; Daughters, R.S.; Lappi, D.A.; Wiley, R.G.; Simone, D.A. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science 1997, 278, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.Q.; Mantyh, P.W.; Carlson, E.J.; Gillespie, A.M.; Epstein, C.J.; Basbaum, A.I. Primary afferent tachykinins are required to experience moderate to intense pain. Nature 1998, 392, 390–394. [Google Scholar] [CrossRef] [PubMed]
- De Felipe, C.; Herrero, J.F.; O’Brien, J.A.; Palmer, J.A.; Doyle, C.A.; Smith, A.J.; Laird, J.M.; Belmonte, C.; Cervero, F.; Hunt, S.P. Altered nociception, analgesia and aggression in mice lacking the receptor for substance P. Nature 1998, 392, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Lin, S.H.; Malewicz, N.M.; Zhang, Y.; Zhang, Y.; Goulding, M.; LaMotte, R.H.; Ma, Q. Identifying the pathways required for coping behaviours associated with sustained pain. Nature 2019, 565, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Rigby, M.; O’Donnell, R.; Rupniak, N.M. Species differences in tachykinin receptor distribution: Further evidence that the substance P (NK1) receptor predominates in human brain. J. Comp. Neurol. 2005, 490, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Gitter, B.D.; Waters, D.C.; Bruns, R.F.; Mason, N.R.; Nixon, J.A.; Howbert, J.J. Species differences in affinities of non-peptide antagonists for substance P receptors. Eur. J. Pharmacol. 1991, 197, 237–238. [Google Scholar] [CrossRef]
- Beresford, I.; Birch, P.; Hagan, R.; Ireland, S. Investigation into species variants in tachykinin NK1 receptors by use of the non-peptide antagonist, CP-96,345. Br. J. Pharmacol. 1991, 104, 292–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appell, K.C.; Fragale, B.J.; Loscig, J.; Singh, S.; Tomczuk, B.E. Antagonists That Demonstrate Species-Differences in Neurokinin-1 Receptors. Mol. Pharmacol. 1992, 41, 772–778. [Google Scholar] [PubMed]
- Mogil, J.S. Animal models of pain: Progress and challenges. Nat. Rev. Neurosci. 2009, 10, 283–294. [Google Scholar] [CrossRef]
- Frederickson, R.C.; Burgis, V.; Harrell, C.E.; Edwards, J.D. Dual actions of substance P on nociception: Possible role of endogenous opioids. Science 1978, 199, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Oehme, P.; Hilse, H.; Morgenstern, E.; Gores, E. Substance-P—Does It Produce Analgesia or Hyperalgesia. Science 1980, 208, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Yoon, T.G.; Kim, S.; Kang, M.; Kim, H.J.; Son, Y. Intravenous Administration of Substance P Attenuates Mechanical Allodynia Following Nerve Injury by Regulating Neuropathic Pain-Related Factors. Biomol. Ther. (Seoul) 2017, 25, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Parenti, C.; Arico, G.; Ronsisvalle, G.; Scoto, G.M. Supraspinal injection of Substance P attenuates allodynia and hyperalgesia in a rat model of inflammatory pain. Peptides 2012, 34, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Bury, R.W.; Mashford, M.L. Biological activity of C-terminal partial sequences of substance P. J. Med. Chem. 1976, 19, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.E.; Stewart, J.M. Substance P and behavior: Opposite effects of N-terminal and C-terminal fragments. Peptides 1983, 4, 763–768. [Google Scholar] [CrossRef]
- Stewart, J.M.; Hall, M.E.; Harkins, J.; Frederickson, R.C.; Terenius, L.; Hokfelt, T.; Krivoy, W.A. A fragment of substance P with specific central activity: SP(1-7). Peptides 1982, 3, 851–857. [Google Scholar] [CrossRef]
- Skilling, S.R.; Smullin, D.H.; Larson, A.A. Differential effects of C- and N-terminal substance P metabolites on the release of amino acid neurotransmitters from the spinal cord: Potential role in nociception. J. Neurosci. 1990, 10, 1309–1318. [Google Scholar] [CrossRef]
- Tang, J.; Chou, J.; Yang, H.Y.; Costa, E. Substance P stimulates the release of Met5-enkephalin-Arg6-Phe7 and Met5-enkephalin from rat spinal cord. Neuropharmacology 1983, 22, 1147–1150. [Google Scholar] [CrossRef]
- Naranjo, J.R.; Arnedo, A.; De Felipe, M.C.; Del Rio, J. Antinociceptive and Met-enkephalin releasing effects of tachykinins and substance P fragments. Peptides 1986, 7, 419–423. [Google Scholar] [CrossRef]
- Naranjo, J.R.; Sanchez-Franco, F.; Garzon, J.; del Rio, J. Analgesic activity of substance P in rats: Apparent mediation by met-enkephalin release. Life Sci. 1982, 30, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, T.; Sasaki, M.; Sanai, K.; Kuwahata, H.; Sakurada, C.; Tsuzuki, M.; Iwata, Y.; Sakurada, S.; Sakurada, T. Intrathecal substance P augments morphine-induced antinociception: Possible relevance in the production of substance P N-terminal fragments. Peptides 2009, 30, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.G.; Yu, L.C. Alterations in the substance P-induced anti-nociception in the central nervous system of rats after morphine tolerance. Neurosci. Lett. 2005, 381, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Bowman, S.L.; Soohoo, A.L.; Shiwarski, D.J.; Schulz, S.; Pradhan, A.A.; Puthenveedu, M.A. Cell-autonomous regulation of Mu-opioid receptor recycling by substance P. Cell Rep. 2015, 10, 1925–1936. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, U.; Nielsen, L.B.; Jensen, K.; Edvinsson, L.; Jansen, I.; Olesen, J. Algesia and local responses induced by neurokinin A and substance P in human skin and temporal muscle. Peptides 1989, 10, 1147–1152. [Google Scholar] [CrossRef]
- Babenko, V.V.; Graven-Nielsen, T.; Svensson, P.; Drewes, A.M.; Jensen, T.S.; Arendt-Nielsen, L. Experimental human muscle pain induced by intramuscular injections of bradykinin, serotonin, and substance P. Eur. J. Pain 1999, 3, 93–102. [Google Scholar] [CrossRef]
- Jensen, K.; Tuxen, C.; Pedersen-Bjergaard, U.; Jansen, I. Pain, tenderness, wheal and flare induced by substance-P, bradykinin and 5-hydroxytryptamine in humans. Cephalalgia 1991, 11, 175–182. [Google Scholar] [CrossRef]
- Lin, C.C.; Chen, W.N.; Chen, C.J.; Lin, Y.W.; Zimmer, A.; Chen, C.C. An antinociceptive role for substance P in acid-induced chronic muscle pain. Proc. Natl. Acad. Sci. USA 2012, 109, E76–E83. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.L.; Cheng, C.F.; Sun, W.H.; Wong, C.W.; Chen, C.C. Targeting ASIC3 for pain, anxiety, and insulin resistance. Pharmacol. Ther. 2012, 134, 127–138. [Google Scholar] [CrossRef]
- Lin, J.H.; Hung, C.H.; Han, D.S.; Chen, S.T.; Lee, C.H.; Sun, W.Z.; Chen, C.C. Sensing acidosis: Nociception or sngception? J. Biomed. Sci. 2018, 25, 85. [Google Scholar] [CrossRef]
- Lin, S.H.; Cheng, Y.R.; Banks, R.W.; Min, M.Y.; Bewick, G.S.; Chen, C.C. Evidence for the involvement of ASIC3 in sensory mechanotransduction in proprioceptors. Nat. Commun. 2016, 7, 11460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.R.; Jiang, B.Y.; Chen, C.C. Acid-sensing ion channels: Dual function proteins for chemo-sensing and mechano-sensing. J. Biomed. Sci. 2018, 25, 46. [Google Scholar] [CrossRef] [PubMed]
- Sluka, K.A.; Kalra, A.; Moore, S.A. Unilateral intramuscular injections of acidic saline produce a bilateral, long-lasting hyperalgesia. Muscle Nerve 2001, 24, 37–46. [Google Scholar] [CrossRef]
- Sluka, K.A.; Price, M.P.; Breese, N.M.; Stucky, C.L.; Wemmie, J.A.; Welsh, M.J. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain 2003, 106, 229–239. [Google Scholar] [CrossRef]
- Chen, W.N.; Chen, C.C. Acid mediates a prolonged antinociception via substance P signaling in acid-induced chronic widespread pain. Mol. Pain 2014, 10, 30. [Google Scholar] [CrossRef]
- Han, D.S.; Lee, C.H.; Shieh, Y.D.; Chen, C.C. Involvement of Substance P in the Analgesic Effect of Low-levle Laser Therapy in a Mouse Model of Chronic Widespread Muscle Pain. Pain Med. 2019. [Google Scholar] [CrossRef]
- Moraes, E.R.; Kushmerick, C.; Naves, L.A. Characteristics of dorsal root ganglia neurons sensitive to Substance P. Mol. Pain 2014, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.Z.; North, R.A. Substance P opens cation channels and closes potassium channels in rat locus coeruleus neurons. Neuroscience 1992, 50, 345–353. [Google Scholar] [CrossRef]
- Nakajima, Y.; Nakajima, S.; Inoue, M. Pertussis toxin-insensitive G protein mediates substance P-induced inhibition of potassium channels in brain neurons. Proc. Natl. Acad. Sci. USA 1988, 85, 3643–3647. [Google Scholar] [CrossRef]
- Takano, K.; Yasufuku-Takano, J.; Kozasa, T.; Singer, W.D.; Nakajima, S.; Nakajima, Y. Gq/11 and PLC-beta 1 mediate the substance P-induced inhibition of an inward rectifier K+ channel in brain neurons. J. Neurophysiol. 1996, 76, 2131–2136. [Google Scholar] [CrossRef]
- Gilbert, R.; Ryan, J.S.; Horackova, M.; Smith, F.M.; Kelly, M.E. Actions of substance P on membrane potential and ionic currents in guinea pig stellate ganglion neurons. Am. J. Physiol. 1998, 274, C892–C903. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.S.; Hille, B. Substance P and somatostatin inhibit calcium channels in rat sympathetic neurons via different G protein pathways. Neuron 1993, 10, 11–20. [Google Scholar] [CrossRef]
- Bley, K.R.; Tsien, R.W. Inhibition of Ca2+ and K+ channels in sympathetic neurons by neuropeptides and other ganglionic transmitters. Neuron 1990, 4, 379–391. [Google Scholar] [CrossRef]
- Lu, B.; Su, Y.; Das, S.; Wang, H.; Wang, Y.; Liu, J.; Ren, D. Peptide neurotransmitters activate a cation channel complex of NALCN and UNC-80. Nature 2009, 457, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.S.; Weinreich, D. Substance P hyperpolarizes vagal sensory neurones of the ferret. J. Physiol. 1996, 493 Pt 1, 157–166. [Google Scholar] [CrossRef]
- Kakehata, S.; Akaike, N.; Takasaka, T. Substance P decreases the non-selective cation channel conductance in dissociated outer hair cells of guinea pig cochlea. Ann. N. Y. Acad. Sci. 1993, 707, 476–479. [Google Scholar] [CrossRef]
- Linley, J.E.; Ooi, L.; Pettinger, L.; Kirton, H.; Boyle, J.P.; Peers, C.; Gamper, N. Reactive oxygen species are second messengers of neurokinin signaling in peripheral sensory neurons. Proc. Natl. Acad. Sci. USA 2012, 109, E1578–E1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Huang, S.; Gao, H.; Liu, Y.; Qi, J.; Chen, P.; Wang, C.; Scragg, J.L.; Vakurov, A.; Peers, C.; et al. Redox-Dependent Modulation of T-Type Ca(2+) Channels in Sensory Neurons Contributes to Acute Anti-Nociceptive Effect of Substance P. Antioxid. Redox Signal. 2016, 25, 233–251. [Google Scholar] [CrossRef]
- Rusin, K.I.; Bleakman, D.; Chard, P.S.; Randic, M.; Miller, R.J. Tachykinins Potentiate N-Methyl-D-Aspartate Responses in Acutely Isolated Neurons from the Dorsal Horn. J. Neurochem. 1993, 60, 952–960. [Google Scholar] [CrossRef]
- Dougherty, P.M.; Willis, W.D. Enhancement of Spinothalamic Neuron Responses to Chemical and Mechanical Stimuli Following Combined Micro-Iontophoretic Application of N-Methyl-d-Aspartic Acid and Substance-P. Pain 1991, 47, 85–93. [Google Scholar] [CrossRef]
- Randic, M.; Hecimovic, H.; Ryu, P.D. Substance P modulates glutamate-induced currents in acutely isolated rat spinal dorsal horn neurones. Neurosci. Lett. 1990, 117, 74–80. [Google Scholar] [CrossRef]
- Rusin, K.I.; Ryu, P.D.; Randic, M. Modulation of Excitatory Amino-Acid Responses in Rat Dorsal Horn Neurons by Tachykinins. J. Neurophysiol. 1992, 68, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.J.; Gover, T.D.; Cordoba-Rodriguez, R.; Weinreich, D. Substance P evokes cation currents through TRP channels in HEK293 cells. J. Neurophysiol. 2003, 90, 2069–2073. [Google Scholar] [CrossRef] [PubMed]
- Cang, C.L.; Zhang, H.; Zhang, Y.Q.; Zhao, Z.Q. PKCepsilon-dependent potentiation of TTX-resistant Nav1.8 current by neurokinin-1 receptor activation in rat dorsal root ganglion neurons. Mol. Pain 2009, 5, 33. [Google Scholar] [CrossRef]
- Zhang, H.; Cang, C.L.; Kawasaki, Y.; Liang, L.L.; Zhang, Y.Q.; Ji, R.R.; Zhao, Z.Q. Neurokinin-1 receptor enhances TRPV1 activity in primary sensory neurons via PKCepsilon: A novel pathway for heat hyperalgesia. J. Neurosci. 2007, 27, 12067–12077. [Google Scholar] [CrossRef] [PubMed]
- Sculptoreanu, A.; Aura Kullmann, F.; de Groat, W.C. Neurokinin 2 receptor-mediated activation of protein kinase C modulates capsaicin responses in DRG neurons from adult rats. Eur. J. Neurosci. 2008, 27, 3171–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapointe, T.K.; Basso, L.; Iftinca, M.C.; Flynn, R.; Chapman, K.; Dietrich, G.; Vergnolle, N.; Altier, C. TRPV1 sensitization mediates postinflammatory visceral pain following acute colitis. Am. J. Physiol.-Gastr. Liver Physiol. 2015, 309, G87–G99. [Google Scholar] [CrossRef] [PubMed]
- Sculptoreanu, A.; de Groat, W.C. Protein kinase C is involved in neurokinin receptor modulation of N- and L-type Ca2+ channels in DRG neurons of the adult rat. J. Neurophysiol. 2003, 90, 21–31. [Google Scholar] [CrossRef]
- Sculptoreanu, A.; Artim, D.E.; de Groat, W.C. Neurokinins inhibit low threshold inactivating K+ currents in capsaicin responsive DRG neurons. Exp. Neurol. 2009, 219, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.K.; Bae, J.H.; Kim, H.Y.; Jo, H.J.; Kim, Y.H.; Jung, S.J.; Kim, J.S.; Oh, S.B. Substance P sensitizes P2X3 in nociceptive trigeminal neurons. J. Dent. Res. 2010, 89, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, T.; Chen, M.; Takeda, D.; King, C.; Ling, J.; Xing, H.; Ataka, T.; Vierck, C.; Yezierski, R.; Gu, J.G. Substance P-driven feed-forward inhibitory activity in the mammalian spinal cord. Mol. Pain 2005, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.S.; Weinreich, D. Substance P regulates Ih via a NK-1 receptor in vagal sensory neurons of the ferret. J. Neurophysiol. 1998, 79, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Lagraize, S.C.; Guo, W.; Yang, K.; Wei, F.; Ren, K.; Dubner, R. Spinal cord mechanisms mediating behavioral hyperalgesia induced by neurokinin-1 tachykinin receptor activation in the rostral ventromedial medulla. Neuroscience 2010, 171, 1341–1356. [Google Scholar] [CrossRef] [PubMed]
- Brink, T.S.; Pacharinsak, C.; Khasabov, S.G.; Beitz, A.J.; Simone, D.A. Differential modulation of neurons in the rostral ventromedial medulla by neurokinin-1 receptors. J. Neurophysiol. 2012, 107, 1210–1221. [Google Scholar] [CrossRef] [PubMed]
- Duggan, A.W.; Hendry, I.A.; Morton, C.R.; Hutchison, W.D.; Zhao, Z.Q. Cutaneous stimuli releasing immunoreactive substance P in the dorsal horn of the cat. Brain Res. 1988, 451, 261–273. [Google Scholar] [CrossRef]
- Debiasi, S.; Rustioni, A. Glutamate and Substance-P Coexist in Primary Afferent Terminals in the Superficial Laminae of Spinal-Cord. Proc. Natl. Acad. Sci. USA 1988, 85, 7820–7824. [Google Scholar] [CrossRef]
- Urban, L.; Randic, M. Slow excitatory transmission in rat dorsal horn: Possible mediation by peptides. Brain Res. 1984, 290, 336–341. [Google Scholar] [CrossRef]
- Wu, L.J.; Xu, H.; Ko, S.W.; Yoshimura, M.; Zhuo, M. Feed-forward inhibition: A novel cellular mechanism for the analgesic effect of substance P. Mol. Pain 2005, 1, 34. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J. Neuronal KCNQ potassium channels: Physiology and role in disease. Nat. Rev. Neurosci. 2000, 1, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Adams, P.R. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 1980, 283, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Mistry, M.; Marsh, S.J.; Brown, D.A.; Delmas, P. Molecular correlates of the M-current in cultured rat hippocampal neurons. J. Physiol. 2002, 544, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passmore, G.M.; Selyanko, A.A.; Mistry, M.; Al-Qatari, M.; Marsh, S.J.; Matthews, E.A.; Dickenson, A.H.; Brown, T.A.; Burbidge, S.A.; Main, M.; et al. KCNQ/M currents in sensory neurons: Significance for pain therapy. J. Neurosci. 2003, 23, 7227–7236. [Google Scholar] [CrossRef]
- Nielsen, A.N.; Mathiesen, C.; Blackburn-Munro, G. Pharmacological characterisation of acid-induced muscle allodynia in rats. Eur. J. Pharmacol. 2004, 487, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Huguenard, J.R. Low-threshold calcium currents in central nervous system neurons. Annu. Rev. Physiol. 1996, 58, 329–348. [Google Scholar] [CrossRef]
- Carbone, E.; Lux, H.D. A Low Voltage-Activated, Fully Inactivating Ca-Channel in Vertebrate Sensory Neurons. Nature 1984, 310, 501–502. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Joksovic, P.M.; Perez-Reyes, E.; Todorovic, S.M. The endogenous redox agent L-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J. Neurosci. 2005, 25, 8766–8775. [Google Scholar] [CrossRef] [PubMed]
- Todorovic, S.M.; Meyenburg, A.; Jevtovic-Todorovic, V. Mechanical and thermal antinociception in rats following systemic administration of mibefradil, a T-type calcium channel blocker. Brain Res 2002, 951, 336–340. [Google Scholar] [CrossRef]
- Berger, N.D.; Gadotti, V.M.; Petrov, R.R.; Chapman, K.; Diaz, P.; Zamponi, G.W. NMP-7 inhibits chronic inflammatory and neuropathic pain via block of Cav3.2 T-type calcium channels and activation of CB2 receptors. Mol. Pain 2014, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, M.F.; Scott, V.E.; McGaraughty, S.; Chu, K.L.; Xu, J.; Niforatos, W.; Milicic, I.; Joshi, S.; Zhang, Q.W.; Xia, Z.R. A peripherally acting, selective T-type calcium channel blocker, ABT-639, effectively reduces nociceptive and neuropathic pain in rats. Biochem. Pharmacol. 2014, 89, 536–544. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Channel Types | Effector System | Cell Types | Species | Effects on Current | Outcomes | References |
|---|---|---|---|---|---|---|
| NMDAR | PKC | Spinal dorsal horn neurons, spinal thalamic neurons | Rat, monkey | ↑ | Pro-nociception | [78,79,80,81] |
| Kir | Gq/11 and PLC-β1 | Locus coeruleus neurons, nucleus basalis neurons | Rat | ↓ | [67,68,69] | |
| KCa | PTX-insensitive G protein | Stellate ganglion neurons | Guinea pig | ↓ | [70] | |
| N-type Ca2+ channel | PTX-insensitive G protein | Stellate ganglion neurons, superior cervical ganglion neurons, sympathetic neurons | Guinea pig, rat, frog | ↓ | [70,71,72] | |
| NALCN | Src family kinases | Hippocampal and ventral tegmental area neurons | Mice | ↑ | [73] | |
| TRP3C | Via NK2R | HEK293 | ↑ | [82] | ||
| Nav1.8 | PKCε | DRG | Rat | ↑ | Pro-nociception | [83] |
| TRPV1 | PKCε | DRG | Rat | ↑ | Pro-nociception | [84,85,86] |
| L, N type calcium channel | PKC | DRG | Rat | ↑ | [87] | |
| Low threshold potassium channel (Kv4) | DRG | Rat | ↓ | Pro-nociception | [88] | |
| P2X3 | TG | Rat | ↑ | Pro-nociception | [89] | |
| GABAAR | Gi/o | Spinal dorsal horn neurons | Rat | ↑ | Anti-nociception | [90] |
| Glycine receptor | Gi/o | Spinal dorsal horn neurons | Rat | ↑ | Anti-nociception | [90] |
| M-type potassium channel | Tyrosine kinase | DRG | Mice | ↑ | Anti-nociception | [57] |
| M-type potassium channel | Gi/o | DRG, TG | Rat | ↑ | Anti-nociception | [76] |
| T-type calcium channel | Gi/o | DRG | Rat | ↓ | Anti-nociception | [77] |
| KCa | Vagal sensory neurons | Ferret | ↑ | [74] | ||
| Ih | Vagal sensory neurons | Ferret | ↓ | [91] | ||
| Non-selective cation channel | PTX-insensitive G protein | Outer hair cells of cochlea | Guinea pig | ↓ | [75] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-T.; Jiang, B.-Y.; Chen, C.-C. Ion Channels Involved in Substance P-Mediated Nociception and Antinociception. Int. J. Mol. Sci. 2019, 20, 1596. https://doi.org/10.3390/ijms20071596
Chang C-T, Jiang B-Y, Chen C-C. Ion Channels Involved in Substance P-Mediated Nociception and Antinociception. International Journal of Molecular Sciences. 2019; 20(7):1596. https://doi.org/10.3390/ijms20071596
Chicago/Turabian StyleChang, Chu-Ting, Bo-Yang Jiang, and Chih-Cheng Chen. 2019. "Ion Channels Involved in Substance P-Mediated Nociception and Antinociception" International Journal of Molecular Sciences 20, no. 7: 1596. https://doi.org/10.3390/ijms20071596
APA StyleChang, C. -T., Jiang, B. -Y., & Chen, C. -C. (2019). Ion Channels Involved in Substance P-Mediated Nociception and Antinociception. International Journal of Molecular Sciences, 20(7), 1596. https://doi.org/10.3390/ijms20071596