Identification of ADAM12 as a Novel Basigin Sheddase

 , and
, and 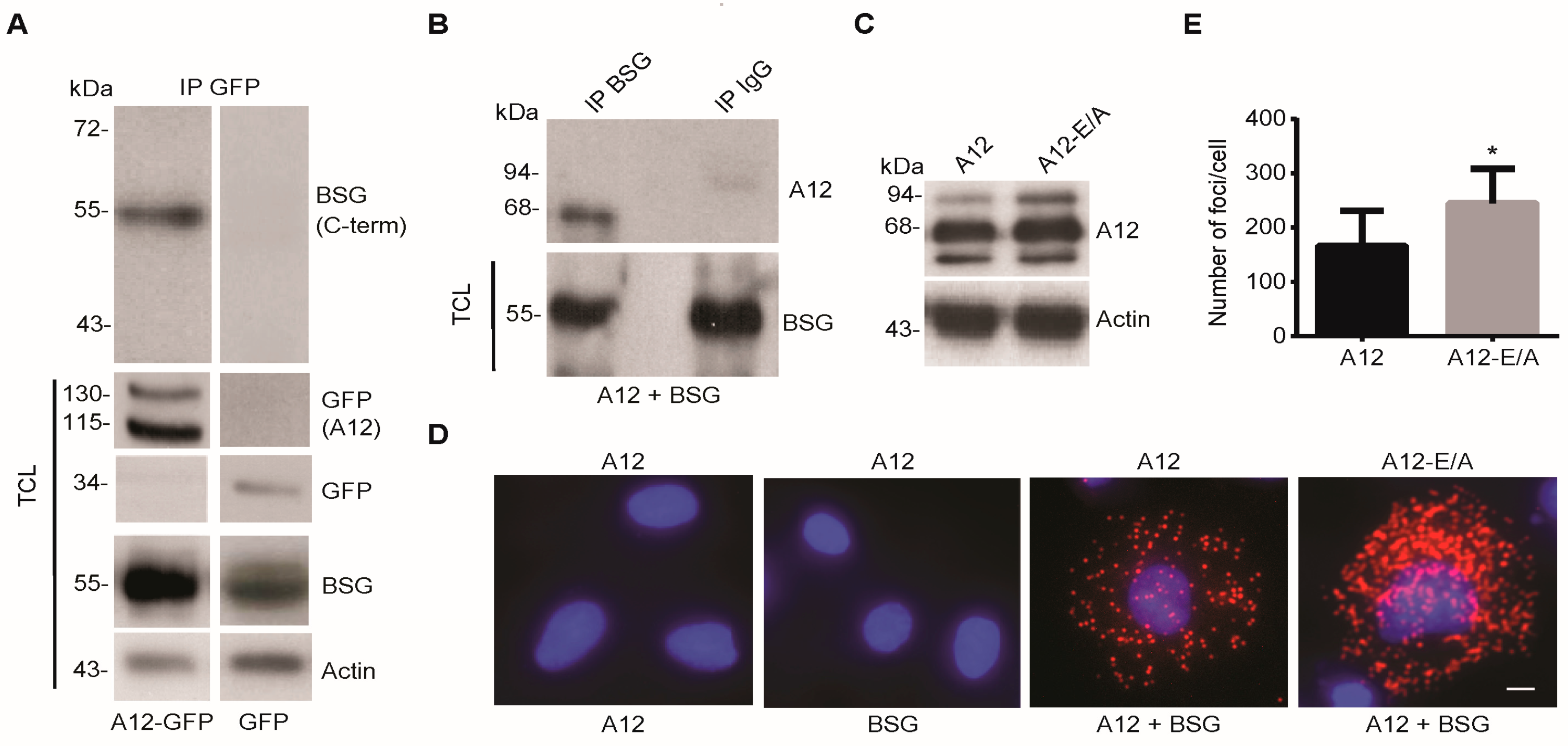{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. ADAM12 Interacts with BSG in Human Cells
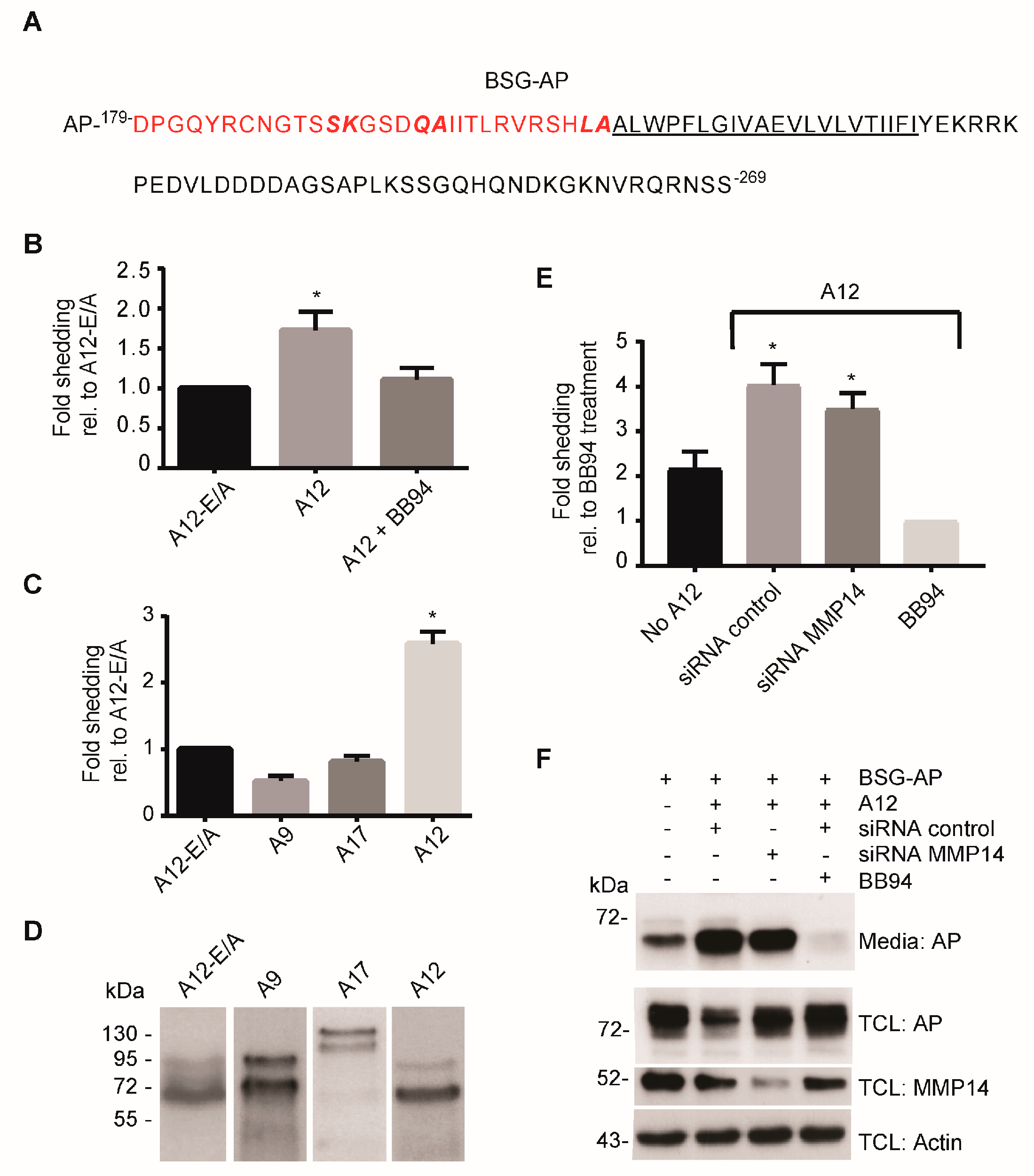
2.2. ADAM12 Overexpression Increases Ectodomain Shedding of a Truncated BSG Reporter Substrate
2.3. CRISPR/Cas9-Mediated ADAM12 Knockout Reduces BSG Reporter Shedding
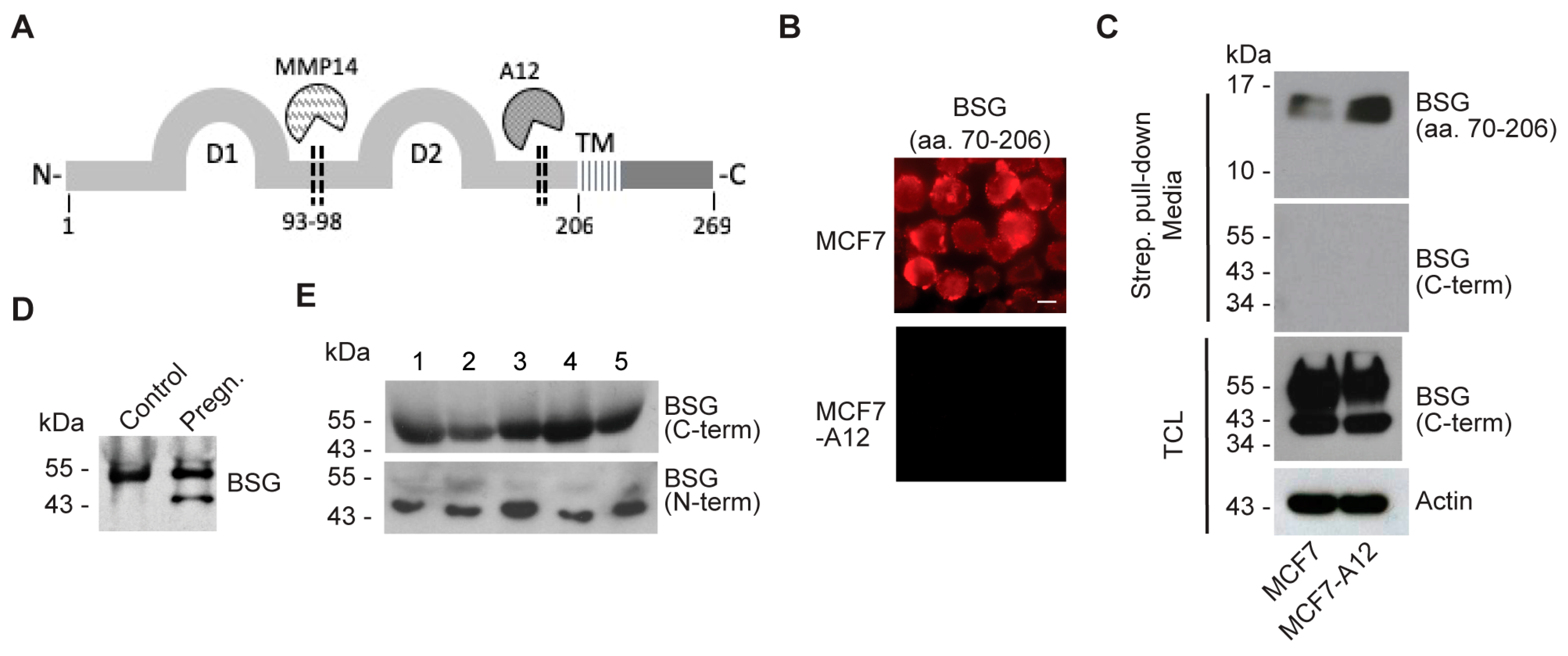
2.4. ADAM12 Sheds Endogenous BSG
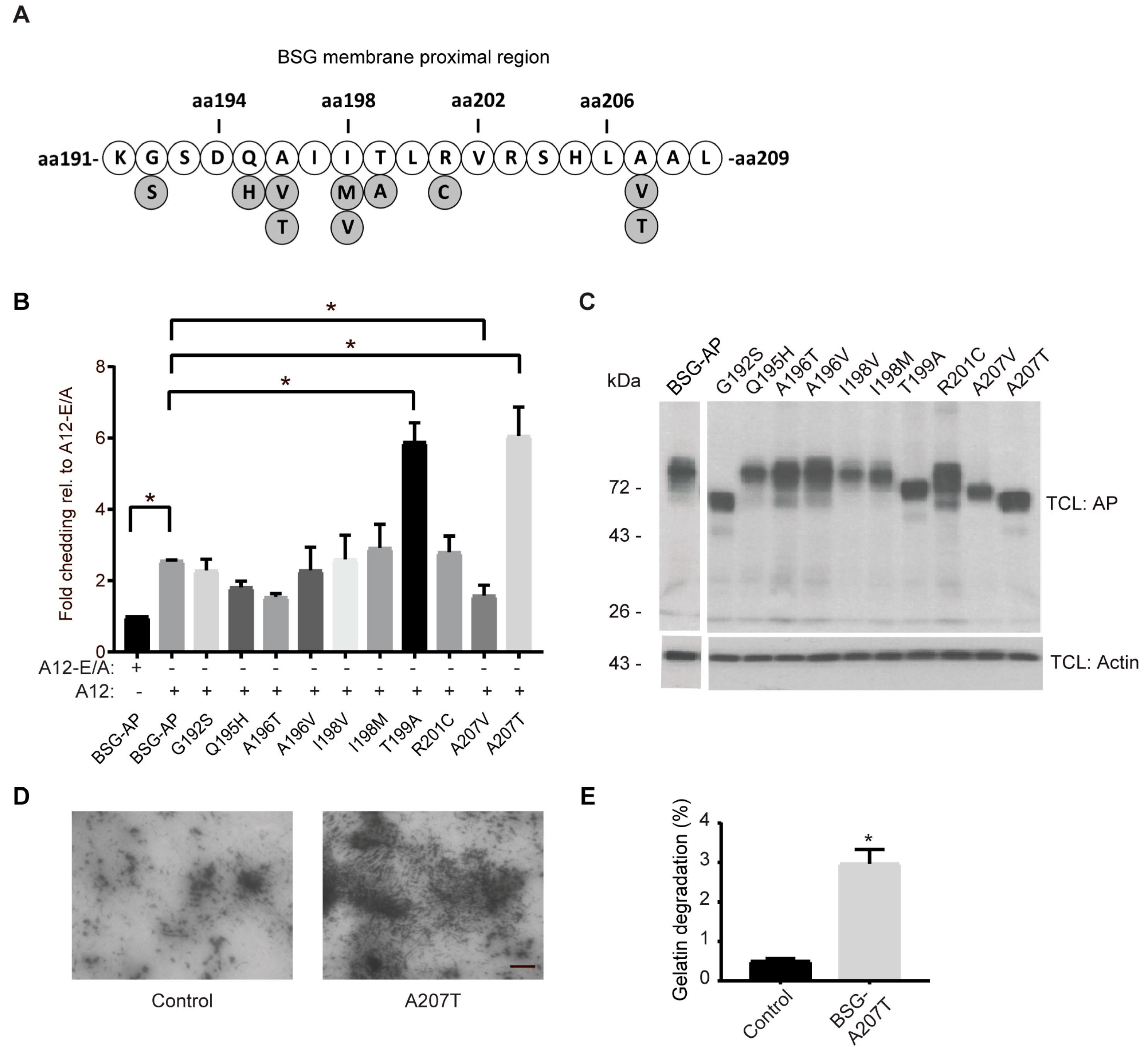
2.5. Cancer-Associated BSG Mutants Are Differentially Shed by ADAM12
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Plasmid
4.3. Cell Culture and Transfections
4.4. CRISPR/Cas9 Gene Editing
4.5. Human Serum Samples
4.6. Shedding Assay
4.7. Immunofluorescence Staining
4.8. In Situ Gelatinase Assay
4.9. Immunoprecipitation and Western Blot Analysis
4.10. Quantitative PCR
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM | A disintegrin and metalloproteinase |
| AP | Alkaline phosphatase |
| BSG | Basigin |
| COSMIC | Catalogue of somatic mutations in cancer |
| ECM | Extracellular matrix |
| EGFR | Epidermal growth factor receptor |
| EMMPRIN | extracellular matrix metalloprotease inducer |
| HRP | Horseradish peroxidase |
| MCT | Monocarboxylate transporter |
| MMP | Matrix metalloproteinase |
| PLA | Proximity ligation assay |
| TNFα | Tumor necrosis factor alpha |
References
- Gabison, E.E.; Hoang-Xuan, T.; Mauviel, A.; Menashi, S. EMMPRIN/CD147, an MMP modulator in cancer, development and tissue repair. Biochimie 2005, 87, 361–368. [Google Scholar] [CrossRef]
- Halestrap, A.P. The SLC16 gene family—Structure, role and regulation in health and disease. Mol. Asp. Med. 2013, 34, 337–349. [Google Scholar] [CrossRef]
- Muramatsu, T. Basigin (CD147), a multifunctional transmembrane glycoprotein with various binding partners. J. Biochem. 2016, 159, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Perez-Escuredo, J.; Van Hee, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 2016, 1863, 2481–2497. [Google Scholar] [CrossRef] [PubMed]
- Kirk, P.; Wilson, M.C.; Heddle, C.; Brown, M.H.; Barclay, A.N.; Halestrap, A.P. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J. 2000, 19, 3896–3904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, C.; Zhang, Y.; DeCastro, R.; Guo, H.; Nakamura, T.; Kataoka, H.; Nabeshima, K. The human tumor cell-derived collagenase stimulatory factor (renamed EMMPRIN) is a member of the immunoglobulin superfamily. Cancer Res. 1995, 55, 434–439. [Google Scholar]
- Gallagher, S.M.; Castorino, J.J.; Wang, D.; Philp, N.J. Monocarboxylate transporter 4 regulates maturation and trafficking of CD147 to the plasma membrane in the metastatic breast cancer cell line MDA-MB-231. Cancer Res. 2007, 67, 4182–4189. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hao, Z.W.; Zhao, Y.X.; Yang, X.M.; Tang, H.; Zhang, X.; Song, F.; Sun, X.X.; Wang, B.; Nan, G.; et al. Full-length soluble CD147 promotes MMP-2 expression and is a potential serological marker in detection of hepatocellular carcinoma. J. Transl. Med. 2014, 12, 190. [Google Scholar] [CrossRef]
- Huang, X.; Shen, W.; Xi, H.; Zhang, K.; Cui, J.; Wei, B.; Chen, L. Prognostic role of extracellular matrix metalloproteinase inducer/CD147 in gastrointestinal cancer: A meta-analysis of related studies. Oncotarget 2016, 7, 81003–81011. [Google Scholar] [CrossRef]
- Bauman, T.M.; Ewald, J.A.; Huang, W.; Ricke, W.A. CD147 expression predicts biochemical recurrence after prostatectomy independent of histologic and pathologic features. BMC Cancer 2015, 15, 549. [Google Scholar] [CrossRef]
- Als, A.B.; Dyrskjot, L.; von der Maase, H.; Koed, K.; Mansilla, F.; Toldbod, H.E.; Jensen, J.L.; Ulhoi, B.P.; Sengelov, L.; Jensen, K.M.; et al. Emmprin and survivin predict response and survival following cisplatin-containing chemotherapy in patients with advanced bladder cancer. Clin. Cancer Res. 2007, 13, 4407–4414. [Google Scholar] [CrossRef]
- Tang, Y.; Kesavan, P.; Nakada, M.T.; Yan, L. Tumor-stroma interaction: Positive feedback regulation of extracellular matrix metalloproteinase inducer (EMMPRIN) expression and matrix metalloproteinase-dependent generation of soluble EMMPRIN. Mol. Cancer Res. 2004, 2, 73–80. [Google Scholar]
- Egawa, N.; Koshikawa, N.; Tomari, T.; Nabeshima, K.; Isobe, T.; Seiki, M. Membrane type 1 matrix metalloproteinase (MT1-MMP/MMP-14) cleaves and releases a 22-kDa extracellular matrix metalloproteinase inducer (EMMPRIN) fragment from tumor cells. J. Biol. Chem. 2006, 281, 37576–37585. [Google Scholar] [CrossRef]
- Wu, B.; Cui, J.; Yang, X.M.; Liu, Z.Y.; Song, F.; Li, L.; Jiang, J.L.; Chen, Z.N. Cytoplasmic fragment of CD147 generated by regulated intramembrane proteolysis contributes to HCC by promoting autophagy. Cell Death Dis. 2017, 8, e2925. [Google Scholar] [CrossRef] [Green Version]
- Murphy, G. The ADAMs: Signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 2008, 8, 929–941. [Google Scholar] [CrossRef]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef] [PubMed]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, K.; Le Gall, S.; Schulte, M.; Yamaguchi, T.; Reiss, K.; Murphy, G.; Toyama, Y.; Hartmann, D.; Saftig, P.; Blobel, C.P. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol. Biol. Cell 2007, 18, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Higashiyama, S.; Nanba, D. ADAM-mediated ectodomain shedding of HB-EGF in receptor cross-talk. Biochim. Biophys. Acta 2005, 1751, 110–117. [Google Scholar] [CrossRef]
- Miller, M.A.; Moss, M.L.; Powell, G.; Petrovich, R.; Edwards, L.; Meyer, A.S.; Griffith, L.G.; Lauffenburger, D.A. Targeting autocrine HB-EGF signaling with specific ADAM12 inhibition using recombinant ADAM12 prodomain. Sci. Rep. 2015, 5, 15150. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, C.; Klitgaard, M.; Noer, J.B.; Kotzsch, A.; Nehammer, C.; Kronqvist, P.; Berthelsen, J.; Blobel, C.; Kveiborg, M.; Albrechtsen, R.; et al. ADAM12 is expressed in the tumour vasculature and mediates ectodomain shedding of several membrane-anchored endothelial proteins. Biochem. J. 2013, 452, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, C.; Nehammer, C.; Albrechtsen, R.; Kronqvist, P.; Kveiborg, M.; Sehara-Fujisawa, A.; Mercurio, A.M.; Wewer, U.M. ADAM12 produced by tumor cells rather than stromal cells accelerates breast tumor progression. Mol. Cancer Res. 2011, 9, 1449–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kveiborg, M.; Frohlich, C.; Albrechtsen, R.; Tischler, V.; Dietrich, N.; Holck, P.; Kronqvist, P.; Rank, F.; Mercurio, A.M.; Wewer, U.M. A role for ADAM12 in breast tumor progression and stromal cell apoptosis. Cancer Res. 2005, 65, 4754–4761. [Google Scholar] [CrossRef] [PubMed]
- Albrechtsen, R.; Kveiborg, M.; Stautz, D.; Vikesa, J.; Noer, J.B.; Kotzsh, A.; Nielsen, F.C.; Wewer, U.M.; Frohlich, C. ADAM12 redistributes and activates MMP-14, resulting in gelatin degradation, reduced apoptosis and increased tumor growth. J. Cell Sci. 2013, 126, 4707–4720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilgin Dogru, E.; Dizdar, Y.; Aksit, E.; Ural, F.; Sanli, O.; Yasasever, V. EMMPRIN and ADAM12 in prostate cancer: Preliminary results of a prospective study. Tumour. Biol. 2014, 35, 11647–11653. [Google Scholar] [CrossRef] [PubMed]
- Kveiborg, M.; Albrechtsen, R.; Couchman, J.R.; Wewer, U.M. Cellular roles of ADAM12 in health and disease. Int. J. Biochem. Cell Biol. 2008, 40, 1685–1702. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, S.; Fredriksson, R.; Wallen-Mackenzie, A. In Situ Proximity Ligation Assay (PLA). Methods Mol. Biol. 2015, 1318, 149–159. [Google Scholar] [CrossRef]
- Kokozidou, M.; Drewlo, S.; Bartz, C.; Raven, G.; Brandenburg, L.O.; Wruck, C.J.; Pufe, T. Complex patterns of ADAM12 mRNA and protein splice variants in the human placenta. Ann. Anat. 2011, 193, 142–148. [Google Scholar] [CrossRef]
- Frohlich, C.; Albrechtsen, R.; Dyrskjot, L.; Rudkjaer, L.; Orntoft, T.F.; Wewer, U.M. Molecular profiling of ADAM12 in human bladder cancer. Clin. Cancer Res. 2006, 12, 7359–7368. [Google Scholar] [CrossRef]
- Moss, M.L.; Rasmussen, F.H. Fluorescent substrates for the proteinases ADAM17, ADAM10, ADAM8, and ADAM12 useful for high-throughput inhibitor screening. Anal. Biochem. 2007, 366, 144–148. [Google Scholar] [CrossRef]
- Taylor, P.M.; Woodfield, R.J.; Hodgkin, M.N.; Pettitt, T.R.; Martin, A.; Kerr, D.J.; Wakelam, M.J. Breast cancer cell-derived EMMPRIN stimulates fibroblast MMP2 release through a phospholipase A(2) and 5-lipoxygenase catalyzed pathway. Oncogene 2002, 21, 5765–5772. [Google Scholar] [CrossRef]
- Knutti, N.; Kuepper, M.; Friedrich, K. Soluble extracellular matrix metalloproteinase inducer (EMMPRIN, EMN) regulates cancer-related cellular functions by homotypic interactions with surface CD147. FEBS J. 2015, 282, 4187–4200. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, M.; Higashi, Y.; Fukushige, T.; Baba, N.; Kawai, K.; Hashiguchi, T.; Su, J.; Zeng, W.; Chen, X.; Kanekura, T. Cleaved CD147 shed from the surface of malignant melanoma cells activates MMP2 produced by fibroblasts. Anticancer Res. 2014, 34, 7091–7096. [Google Scholar]
- Belton, R.J., Jr.; Chen, L.; Mesquita, F.S.; Nowak, R.A. Basigin-2 is a cell surface receptor for soluble basigin ligand. J. Biol. Chem. 2008, 283, 17805–17814. [Google Scholar] [CrossRef]
- Yu, X.L.; Hu, T.; Du, J.M.; Ding, J.P.; Yang, X.M.; Zhang, J.; Yang, B.; Shen, X.; Zhang, Z.; Zhong, W.D.; et al. Crystal structure of HAb18G/CD147: Implications for immunoglobulin superfamily homophilic adhesion. J. Biol. Chem. 2008, 283, 18056–18065. [Google Scholar] [CrossRef]
- Albrechtsen, R.; Stautz, D.; Sanjay, A.; Kveiborg, M.; Wewer, U.M. Extracellular engagement of ADAM12 induces clusters of invadopodia with localized ectodomain shedding activity. Exp. Cell Res. 2011, 317, 195–209. [Google Scholar] [CrossRef]
- Iba, K.; Albrechtsen, R.; Gilpin, B.J.; Loechel, F.; Wewer, U.M. Cysteine-rich domain of human ADAM 12 (meltrin alpha) supports tumor cell adhesion. Am. J. Pathol. 1999, 154, 1489–1501. [Google Scholar] [CrossRef]
- Hansson, M.D.; Rzeznicka, K.; Rosenback, M.; Hansson, M.; Sirijovski, N. PCR-mediated deletion of plasmid DNA. Anal. Biochem. 2008, 375, 373–375. [Google Scholar] [CrossRef]
- Sanjay, A.; Houghton, A.; Neff, L.; DiDomenico, E.; Bardelay, C.; Antoine, E.; Levy, J.; Gailit, J.; Bowtell, D.; Horne, W.C.; et al. Cbl associates with Pyk2 and Src to regulate Src kinase activity, alpha(v)beta(3) integrin-mediated signaling, cell adhesion, and osteoclast motility. J. Cell Biol. 2001, 152, 181–195. [Google Scholar] [CrossRef]
- Ronnov-Jessen, L.; Villadsen, R.; Edwards, J.C.; Petersen, O.W. Differential expression of a chloride intracellular channel gene, CLIC4, in transforming growth factor-beta1-mediated conversion of fibroblasts to myofibroblasts. Am. J. Pathol. 2002, 161, 471–480. [Google Scholar] [CrossRef]
- Hodgkins, A.; Farne, A.; Perera, S.; Grego, T.; Parry-Smith, D.J.; Skarnes, W.C.; Iyer, V. WGE: A CRISPR database for genome engineering. Bioinformatics 2015, 31, 3078–3080. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, N.; Sundberg, C.; Kveiborg, M.; Moghadaszadeh, B.; Asmar, M.; Dietrich, N.; Thodeti, C.K.; Nielsen, F.C.; Moller, P.; Mercurio, A.M.; et al. ADAM12 induces actin cytoskeleton and extracellular matrix reorganization during early adipocyte differentiation by regulating beta1 integrin function. J. Cell Sci. 2003, 116, 3893–3904. [Google Scholar] [CrossRef] [PubMed]
- Stautz, D.; Leyme, A.; Grandal, M.V.; Albrechtsen, R.; van Deurs, B.; Wewer, U.; Kveiborg, M. Cell-surface metalloprotease ADAM12 is internalized by a clathrin- and Grb2-dependent mechanism. Traffic 2012, 13, 1532–1546. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albrechtsen, R.; Wewer Albrechtsen, N.J.; Gnosa, S.; Schwarz, J.; Dyrskjøt, L.; Kveiborg, M. Identification of ADAM12 as a Novel Basigin Sheddase. Int. J. Mol. Sci. 2019, 20, 1957. https://doi.org/10.3390/ijms20081957
Albrechtsen R, Wewer Albrechtsen NJ, Gnosa S, Schwarz J, Dyrskjøt L, Kveiborg M. Identification of ADAM12 as a Novel Basigin Sheddase. International Journal of Molecular Sciences. 2019; 20(8):1957. https://doi.org/10.3390/ijms20081957
Chicago/Turabian StyleAlbrechtsen, Reidar, Nicolai J. Wewer Albrechtsen, Sebastian Gnosa, Jeanette Schwarz, Lars Dyrskjøt, and Marie Kveiborg. 2019. "Identification of ADAM12 as a Novel Basigin Sheddase" International Journal of Molecular Sciences 20, no. 8: 1957. https://doi.org/10.3390/ijms20081957
APA StyleAlbrechtsen, R., Wewer Albrechtsen, N. J., Gnosa, S., Schwarz, J., Dyrskjøt, L., & Kveiborg, M. (2019). Identification of ADAM12 as a Novel Basigin Sheddase. International Journal of Molecular Sciences, 20(8), 1957. https://doi.org/10.3390/ijms20081957