1. Introduction
Osteocytes are the major cell type in the bone and account for up to 95% of bone cells [
1]. They are located in mineralized bone and are in contact with osteoblasts on the bone surface via gap junctions [
2]. The poor accessibility of osteocytes and their difficult cultivation have only recently made it possible to develop a suitable isolation method [
3] and in vitro environment [
4]. It has been supposed, that osteocytes orchestrate bone homeostasis by regulating bone-forming osteoblasts and bone-resorbing osteoclasts. Interactions between osteoblasts and osteocytes have already been demonstrated: mature osteocytes express sclerostin and Dkk1, which have a negative effect on the Wnt/β-catenin pathway. This pathway plays an important role in the regulation of bone mass by osteoblasts and its inhibition suppresses the activity of osteoblasts [
5,
6,
7]. This relationship is confirmed by sclerostin (SOST) knockout mice and Dkk mutant mice, which showed a high bone mass with increased bone formation [
8,
9].
In vitro co-culture of osteocytes and osteoblasts are an additional approach to study the interaction of these cell types. Some studies on in vitro co-cultivation were already published. Taylor and co-workers cultivated osteocytes and osteoblasts on different sides of a transwell insert membrane [
10], however 2D cultivation of osteocytes does not imitate the situation in vivo, where osteocytes are embedded into the 3D bone matrix. Other groups embedded osteocytes in collagen gels and seeded osteoblasts on top of this construct [
11,
12]. The missing spatial separation of the two cell types and the potential of osteoblasts to migrate into collagen gels can cause cross-contamination of the RNA between osteocytes and osteoblasts, which makes separate gene expression analysis impossible. In addition, all published co-culture studies used cell lines for osteoblasts and osteocytes (e.g., MLO-Y4 and MC3T3-E1) although immortalized cell lines do not fully resemble the behavior of primary cells due to phenotypic and genetic differences [
3,
13]. Therefore, an in vitro co-culture model of osteoblasts and osteocytes, comprising solely human primary cells would be highly advantageous. Such in vitro co-cultures can be used for further investigations of osteocyte-osteoblast interaction. Additionally, the effect of growth factors or drugs on the bone cell interaction can be studied.
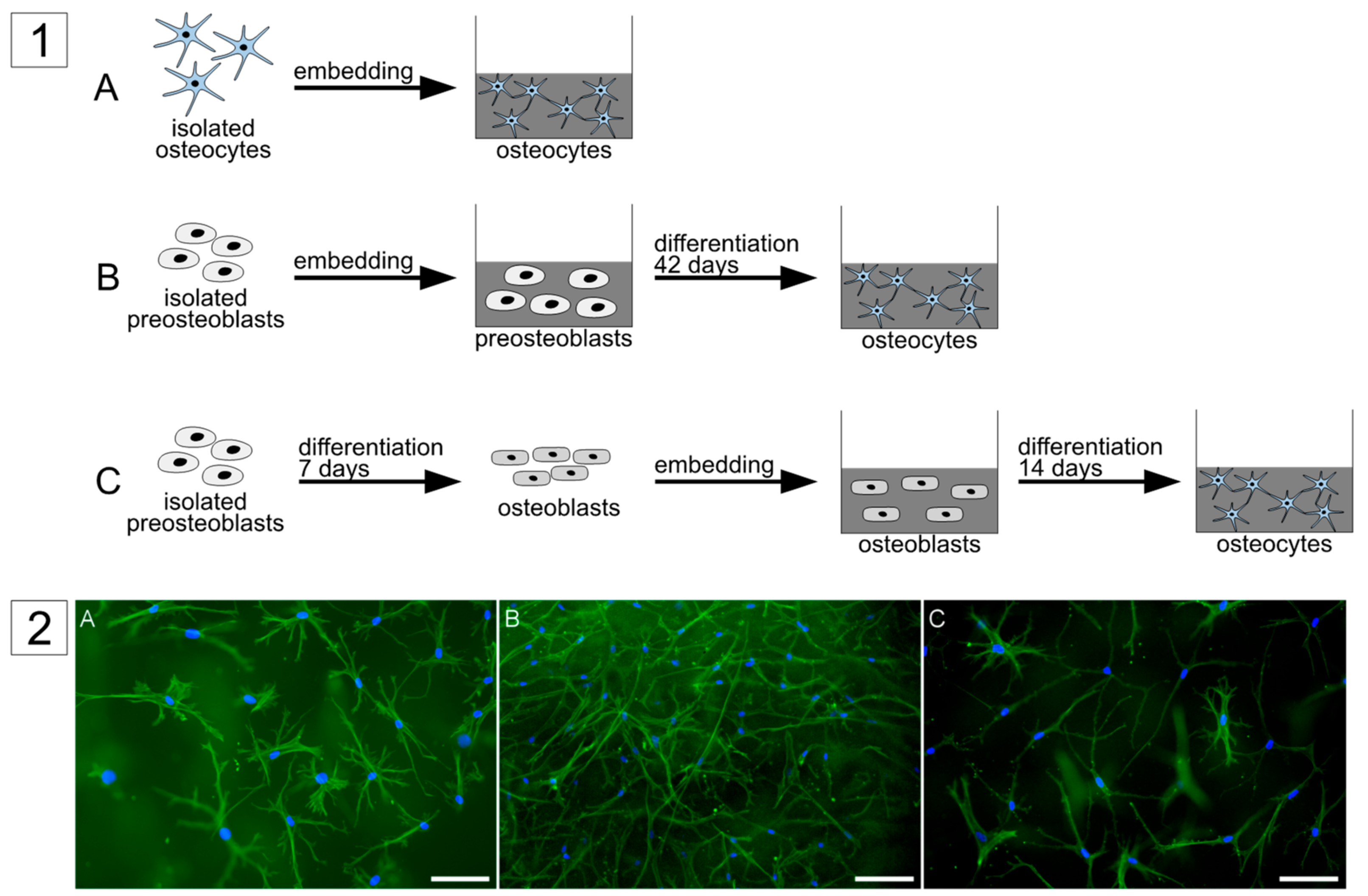
The main aim of this study was to develop a functional co-culture with suitable conditions for osteocytes and osteoblasts and to identify interactions between the two cell types. Therefore, osteocytes were cultivated in a 3D collagen matrix to ensure a natural bone environment. Three different approaches to obtain human primary osteocytes were compared, to find the optimal approach for co-cultivation: (i) direct isolation of primary human osteocytes from bone tissue by sequential digestion and demineralization, (ii) long-term differentiation of pre-osteoblasts in collagen gels, and (iii) short time differentiation of mature osteoblasts in collagen gels. The three types of osteocyte cultures were embedded in collagen gels, which were inserted into commercially available transwell inserts. Mature osteoblasts were seeded onto the basal side of the porous transwell insert membrane, enabling the separation of osteocytes and osteoblasts for analysis. In all co-culture experiments, primary osteoblasts and osteocytes derived from the same donor to imitate the conditions in human bone.
3. Discussion
In this paper, a co-culture of primary osteocytes and osteoblasts was established, to investigate the interaction of these bone cell types. Some challenges have prevented the development of such co-cultures so far: primary osteocytes are difficult to culture, because they need a 3D collagen matrix in vitro to form dendritic extensions and maintain their osteocytic phenotype [
4]. Adding osteoblasts onto the surface of collagen gels results in a migration process of the osteoblasts from the surface into the gel and simultaneous differentiation to osteocytes [
12,
14]. Therefore, a spatial separation from the osteoblasts to the collagen gel is necessary. Additionally, osteocyte cultivation requires a medium with low fetal calf serum (FCS) [
3,
15], while the common medium for osteoblasts contains 10% FCS. Thus, a medium supporting the differentiation of both cell types had to be found. It has been shown that the cultivation of osteoblasts under low FCS concentrations promotes the osteogenic phenotype [
16]. Therefore, medium containing 2% FCS was suitable for both cell types in the co-culture.
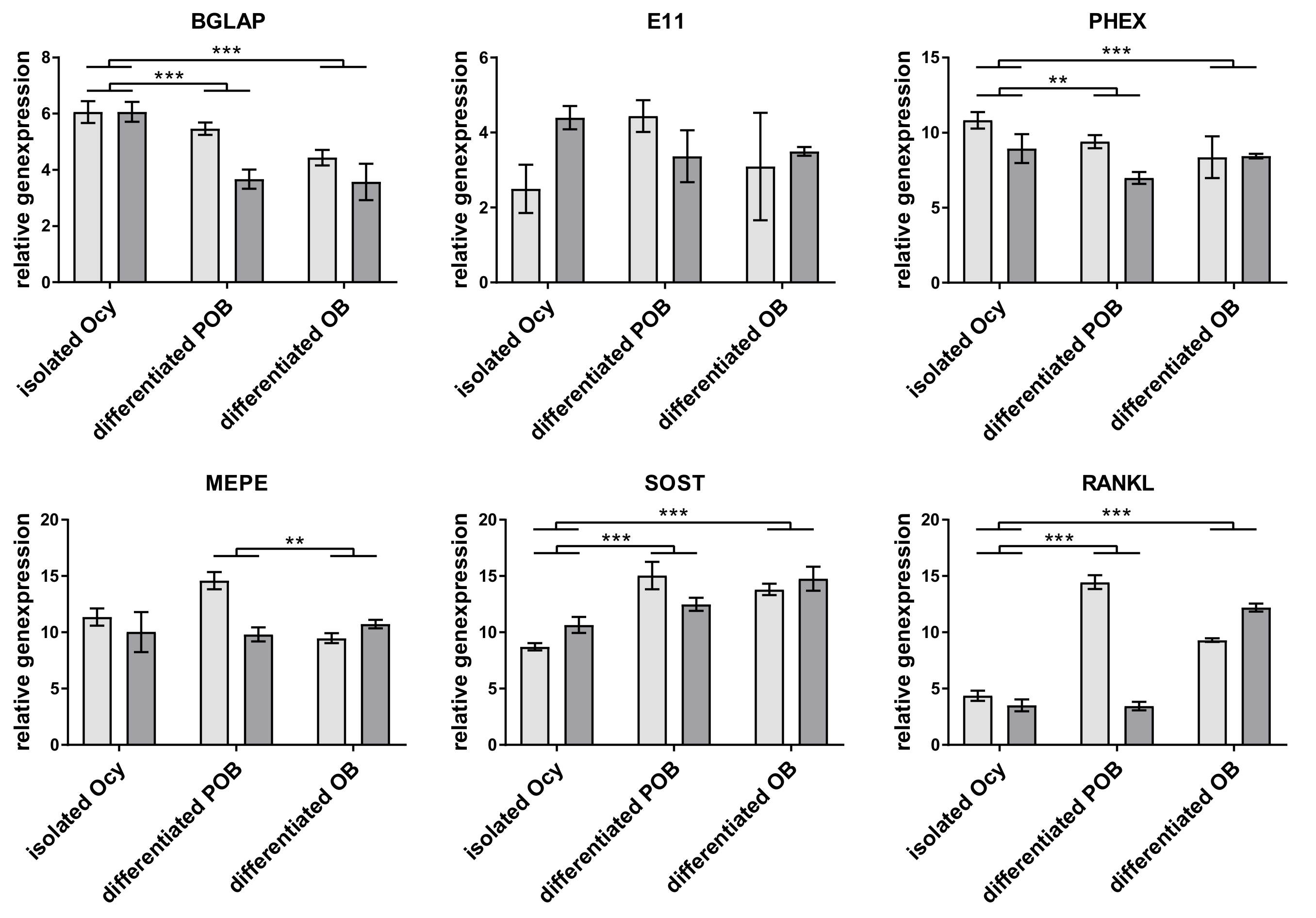
Current in vitro studies on osteocyte-osteoblast interaction were invariably performed with rodent cells, and the present study is the first to establish an in vitro co-culture model comprising solely primary human osteocytes and osteoblasts. One of the aims of this study was to identify suitable conditions for the generation of human primary osteocytes. Therefore, we compared three approaches: (i) direct isolation of osteocytes from bone tissue by multistep digestion, (ii) long-time differentiation of human pre-osteoblasts embedded in collagen gels, and (iii) short-time differentiation of mature human osteoblasts in collagen gels. With all approaches it was possible to obtain differentiated dendritic osteocytes expressing osteocytic marker including SOST, which cannot be observed in the commonly used MYO-Y4 osteocytes [
17].
To decide on an approach suitable for co-culture experiments, the time factor and the gained cell amount play an important role. Since the multi-step digestion of bone to receive primary osteocytes is very time consuming and only a few cells are obtained, the first method is not very efficient. The second approach needs a long period of six weeks differentiation from pre-osteoblasts to osteocytes. During the first period of differentiation, the cells proliferate, resulting in a high final osteocyte number in the gels. Those high osteocyte numbers bear the risk of gel shrinking [
4]. Furthermore, the osteocyte morphology might be more difficult to evaluate in gels with a high cell density. The last approach with differentiation of mature osteoblasts just needs three weeks including pre-differentiation of the osteoblasts and is therefore suggested as the easiest, fastest, and most efficient approach to obtain primary osteocytes.
Although there are already a few approaches for co-cultures of osteocytes and osteoblasts [
10,
11,
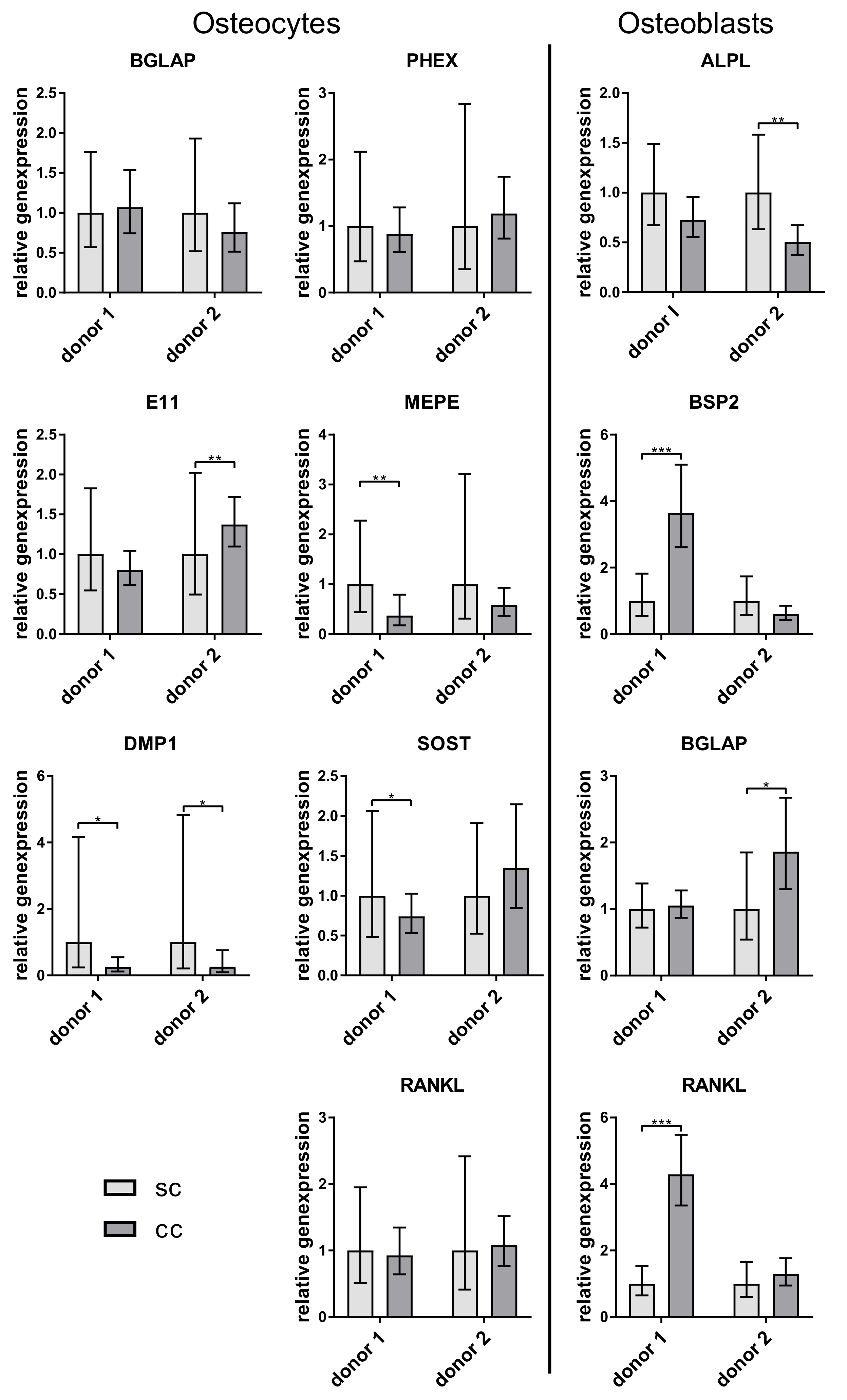
12], differences between single and co-cultures of osteocytes and osteoblasts have not been examined. Our study showed a stable expression of osteocyte markers both in single and co-culture. All osteocytes expressed sclerostin, a protein that was shown to inhibit the osteogenic differentiation and the function of the osteoblasts [
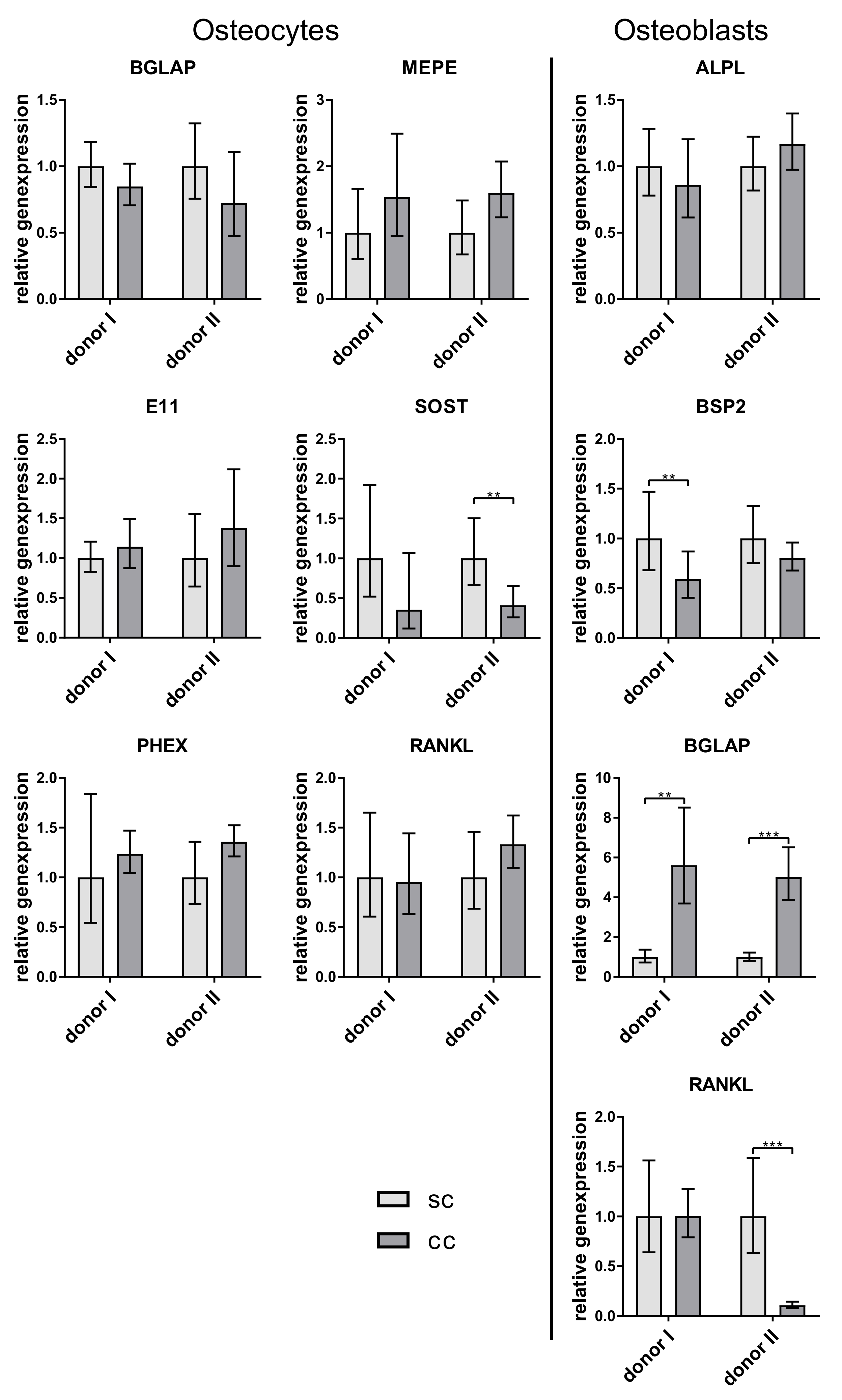
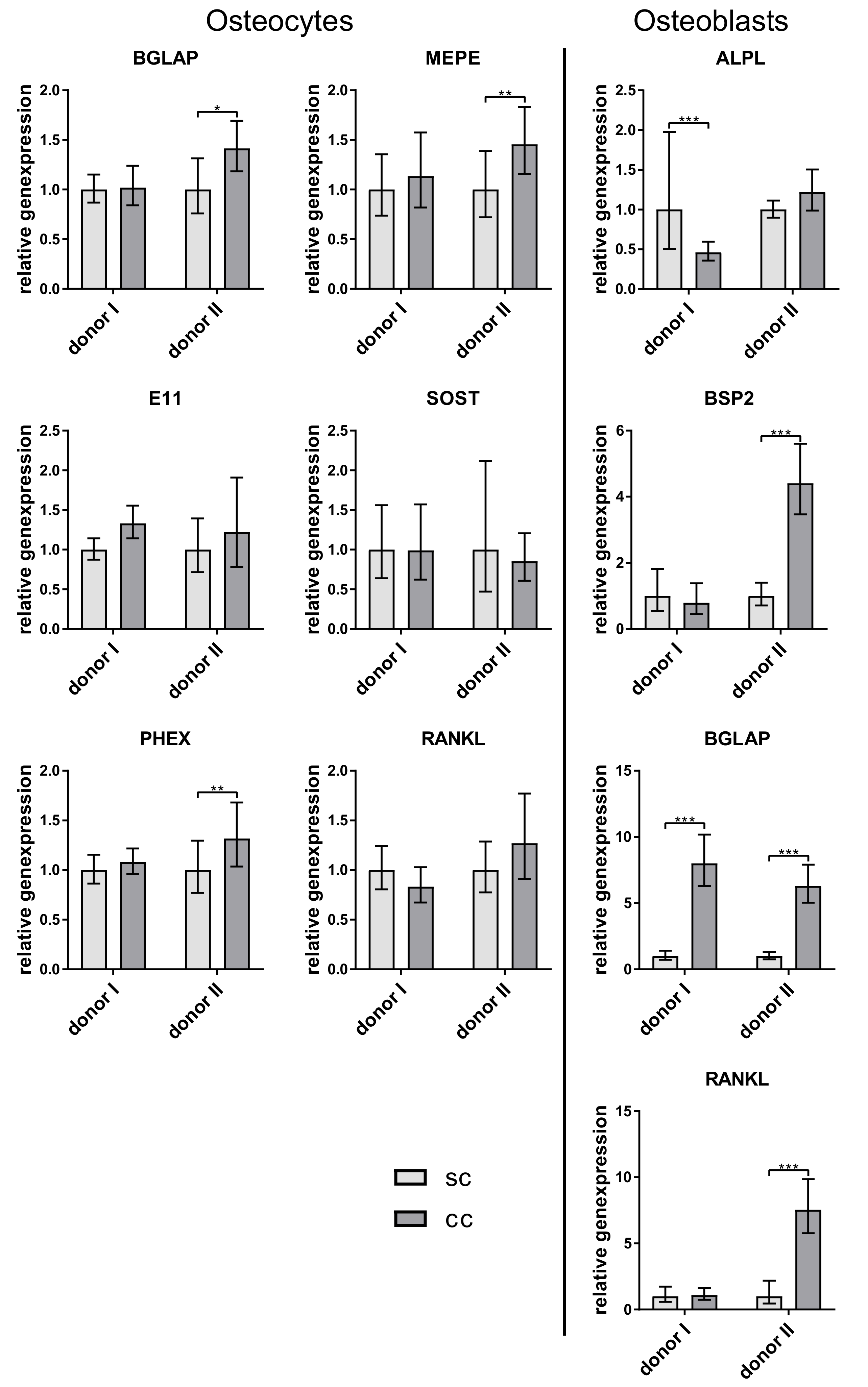
6]. However, in the co-culture experiments of our study, no downregulation of the examined osteoblast markers ALPL, BSPII, and RANKL was observed. Probably the amount of sclerostin which was synthesized by the osteocytes in vitro was too low to have an effect on the osteoblasts. Further studies should involve the quantification of SOST not only on gene but also on protein level.
RANKL is expressed by osteocytes and osteoblasts and promotes osteoclastogenesis [
18]. It has been shown for murine cells, that osteocytes express even more RANKL than osteoblasts [
19]. In our experiments, there was no clearly higher RANKL expression in osteocytes. We detected both higher and lower RANKL expression in osteocytes compared to osteoblasts of the same experiment (data not shown). However, freshly isolated osteocytes showed the highest RANKL expression (
Figure 2). A very interesting observation of our co-culture study was that osteoblasts expressed significantly more osteocalcin when cultivated with osteocytes. This is the first time that a stimulation of osteoblasts by osteocytes, which were not mechanically treated before, was shown in vitro. Regulatory elements in the promoter of the osteocalcin gene were already characterized, indicating a strong transcriptional regulation of this gene. This regulation is mainly controlled by transcription factors like Runx2, AP-1 related proteins, or homeobox proteins [
20].
Several in vitro studies indicated that Runx2 (Cbfa1/AML3) is a positive regulator of osteocalcin expression at an early stage of osteoblast maturation [
21,
22]. While at a late stage of maturation, the overexpression of Runx2 in osteoblasts failed to upregulate osteocalcin expression. This indicates that some other factors are required for the regulation of osteocalcin expression at the later stages [
22]. Since we used mature osteoblasts for co-cultures, the increase in osteocalcin expression may not be caused by a regulation of the Runx2 transcription factor. AP-1 is a transcription factor that binds DNA by forming a heterodimer composed of proteins belonging to the c-Fos and c-Jun families (c-Jun, JunB and JunD, c-Fos, FosB, Fra-1, and Fra-2) [
23]. Grigoriadis et al. demonstrated that osteoblastic cells are principal targets for c-Fos [
24]. However, they observed a reduction of osteocalcin expression in cell lines with high c-fos expression. Mason et al. proved that normal and mechanically stimulated osteocytes in the cortical bone of ulnae of rats constitutively express c-Fos and c-Jun [
25]. These data suggest that c-Fos and c-Jun, expressed by osteocytes, might be involved in the gene regulation of osteocalcin in osteoblasts. Homebox proteins (Msx1, Msx2, Dlx5, and Dlx6) play a role in osteoblast differentiation and osteocalcin expression, too. Yet, an effect of dlx3 and dlx5 on the osteocalcin gene expression has been demonstrated [
23].
A regulation of these transcription factors by osteocytes is possible by their release of growth factors like BMP2, TGFβ, IGF-1, PGE2, FGF-2, and others. Their effect has mainly been studied on the Runx2transcription factor, but BMP2 also promotes Dlx5 expression in osteoblasts, which activates osteocalcin expression [
23].
The function of osteocalcin in the co-culture could not yet be investigated, but various studies demonstrated that osteocalcin is a regulator of mineralization by binding calcium ions [
26].
In contrast to the effects of osteocytes on osteoblast osteocalcin expression, the present study did not show any significant effects of osteoblasts on osteocyte gene expression. This observation may suggest, that possible interactive effects of osteoblasts on osteocytes are negligible, however, it is still possible that those effects exist in a narrow zone of osteocytes, which is in close proximity to the osteoblast layer.
In summary, it can be concluded that osteocytes stimulate osteocalcin expression in osteoblasts even without mechanical stimulation or external factors. However, this stimulation could be further enhanced by such additional factors, which can be further investigated. The regulation of the osteocalcin gene by osteocytes is a very complex mechanism, involving many different factors. Because osteocalcin has skeletal and metabolic functions [
26], further research for a better understanding of this regulation is necessary, to offer new therapeutic strategies against bone diseases and metabolic disorders. The co-culture established is this paper is suitable for such studies.
4. Materials and Methods
4.1. Isolation of Primary Human Osteocytes and Pre-Osteoblasts
Osteocytes and pre-osteoblasts were isolated from human femoral heads of osteoarthritic patients undergoing total hip replacement at the University Hospital Carl Gustav Carus Dresden (Germany) after informed consent (approval by the ethics commission of TU Dresden, EK 3030814, 8. August. 2014). For osteocyte isolation bone material of two donors was used (donor 1: female 67 years, donor 2: male 77 years). For pre-osteoblast isolation bone material of the above mentioned two donors, and additionally, from another two donors was used (donor I: female 56 years, donor II: female 75 years). Osteocyte isolation was performed based on a protocol of Prideaux and co-workers [
3] with some modifications (Bernhardt et al. Biomedical Engineering, in press). Spongious bone fragments (1–2 mm) were repeatedly digested (collagenase II treatment) and demineralized (EDTA treatment). Resting steps of the particles in between the treatments increased the yield of osteocytes. Isolated primary osteocytes after at least six digestion steps were cultivated on collagen-coated TCPS for 2 days with α-MEM, 2% fetal calf serum (FCS), 2 mM L-glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin, before embedding the cells into collagen gels. Pre-osteoblasts were isolated from spongious bone fragments (1–2 mm) after two collagenase digestions. Cells were expanded in α-MEM, 15% FCS, 2 mM L-glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin, before use in the different experiments.
4.2. Differentiation of Pre-Osteoblasts
Pre-osteoblasts were differentiated in vitro for 7 days in α-MEM, 10% FCS, 10 mM β-glycerophosphate, 50 µM ascorbic acid 2-phosphate, 10−7 M dexamethasone, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin to get mature osteoblasts.
4.3. Preparation of Collagen Gels
Collagen gels were prepared by mixing 9 parts of acidic collagen solution (3 mg/mL rat collagen in 0.1 M acetic acid, Amedrix, Esslingen, Germany) with 1 part of 10× HBSS and neutralizing with 1 M NaOH. For embedding of osteocytes, pre-osteoblasts or osteoblasts the respective cell suspension was added to the neutralized collagen at a concentration of 1 × 105 cells/mL. Cell laden collagen solution (0.5 mL) was added to 12-well transwell inserts with 0.4 µm pore size.
4.4. Co-Culture
Transwell inserts, filled with cell-laden collagen gels (osteocytes, directly isolated from bone or differentiated from osteoblasts, according to the respective approach) were inverted to add 1 × 105 pretreated osteoblasts (always from the same donor as the osteocytes) onto the basal side of the membrane, and incubated for 4 h at 37 °C to allow cellular adhesion. Afterwards the inserts were reverted and the constructs were cultivated in α-MEM, 2% FCS, 50 µM ascorbic acid 2-phosphate, 10 mM β-glycerophosphate, 2 mM L-glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin. Additionally, single cultures of osteocytes, embedded in collagen gels in transwell inserts and single cultures of osteoblasts on the basal side of transwell inserts, which were filled with cell-free collagen gels were performed. Three replicates were used per culture and for every replicate, two samples were pooled.
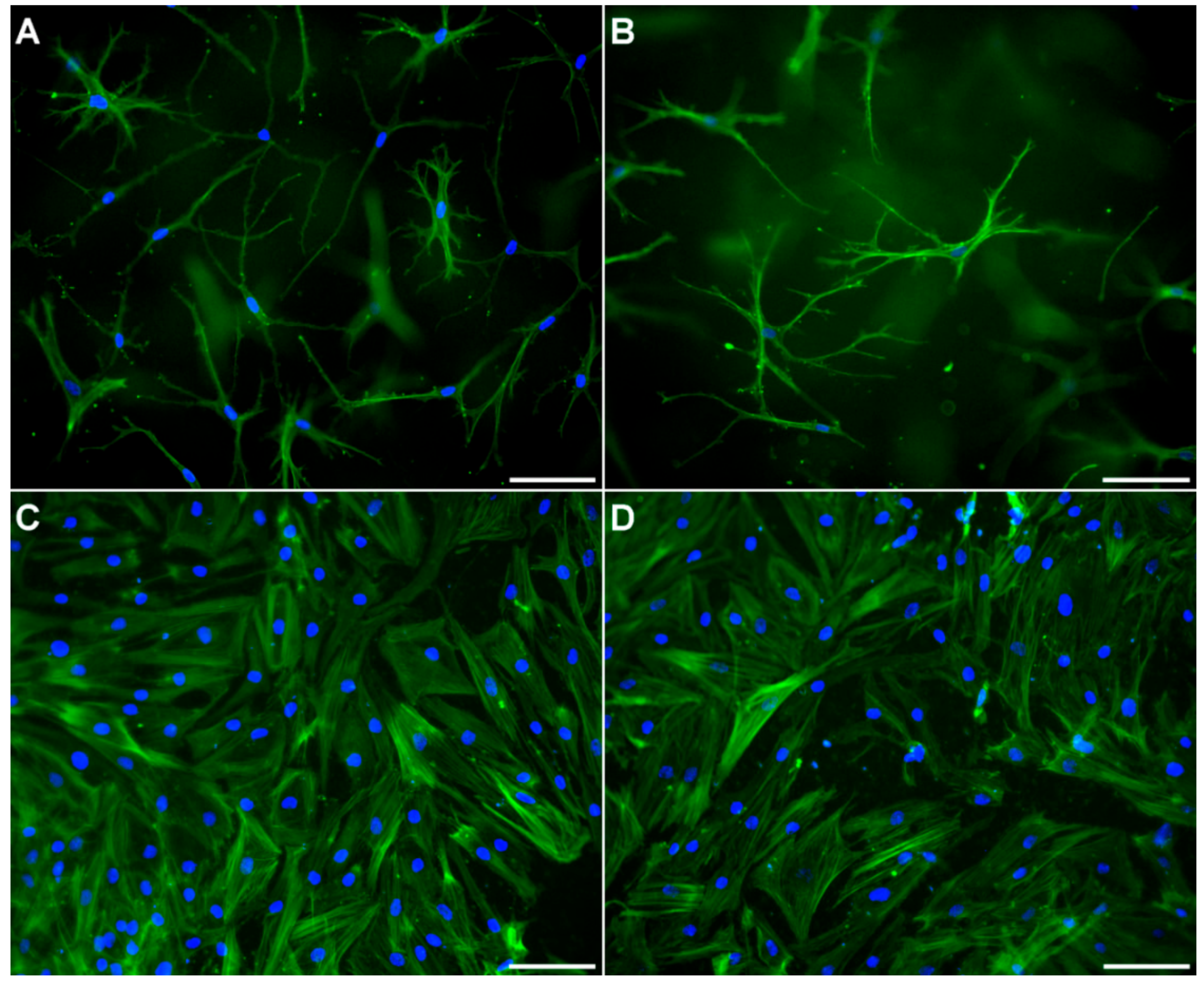
4.5. Fluorescence Microscopy
Osteocyte-containing gels were removed from the transwell inserts and osteoblast-seeded membranes were cut out of the insert with a scalpel. Gels and membranes were fixed separately with a 4% solution of neutral buffered formaldehyde for 1 h at RT. After permeabilization with 0.1% Triton X-100 in PBS for 5 min and 6 washes with PBS, samples were blocked using 3% bovine serum albumin in PBS for 30 min. The samples were further incubated with a solution of 20 ng/mL DAPI (4′,6-diamidin-2-phenylindol, Invitrogen) and 25 µL/mL AlexaFluor 488® phalloidin (Invitrogen, part of Thermo Fisher Scientific, Waltham, MA, USA) overnight at 4 °C protected from light. After removal of the staining solution, the samples were finally washed with PBS and imaged with a Keyence BZ 9000 fluorescence microscope.
4.6. RNA Isolation and cDNA Synthesis
Cell-seeded collagen gels (six 0.5 mL-gels per experimental group) were removed from the transwell inserts and incubated with collagenase II solution (3 mg/mL collagenase II, 235 U/mg, Biochrom, Berlin, Germany in α-MEM, 10% FCS, 2 mM L-glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin, 3 mM CaCl2) for 1 h at 37 °C. The digests were transferred to 15 mL tubes, washed with PBS and centrifuged. The pellets were washed with PBS and RNA was isolated from these pellets as well as from the osteoblast-seeded transwell membranes using a commercially available kit (peqGOLD MicroSpin Total RNA Kit, Peqlab, Erlangen, Germany). cDNA was generated using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, part of Thermo Fisher Scientific) according to manufacturer’s instructions.
4.7. PCR
PCR reactions were set up using the TaqMan Fast Advanced Master Mix (Thermo Fisher) and TaqMan Gene Expression Assays for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), bone gamma-carboxyglutamate protein (osteocalcin, BGLAP), podoplanin (E11/g38; PDPN), phosphate regulating endopeptidase homolog, X-linked (PHEX), matrix extracellular phosphoglycoprotein (OPF 45, MEPE), receptor activator of NF-κB Ligand (RANKL, TNFSF11), dentin matrix protein 1 (DMP-1), sclerostin (SOST), alkaline phosphatase (ALPL), and bone sialoprotein II (BSP II) (Applied Biosystems, part of Thermo Fisher Scientific) according to manufacturer’s instructions.
PCR was run with an Applied Biosystems® 7500 fast Real-Time PCR system (Applied Biosystems, part of Thermo Fisher Scientific). Relative gene expression (fold-change) was calculated using the 2−ΔΔCt method and normalized to GAPDH expression. For the comparison of different approaches to generate primary human osteocytes, the ΔCT values were directly compared.
Statistical differences of different groups were calculated at the level of ΔCT values using one-way or two-way ANOVA followed by Post hoc Tukey test to determine multiple comparisons (Origin 9.1, OriginLab).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





