Citrate-Induced p85α–PTEN Complex Formation Causes G2/M Phase Arrest in Human Pharyngeal Squamous Carcinoma Cell Lines



Abstract
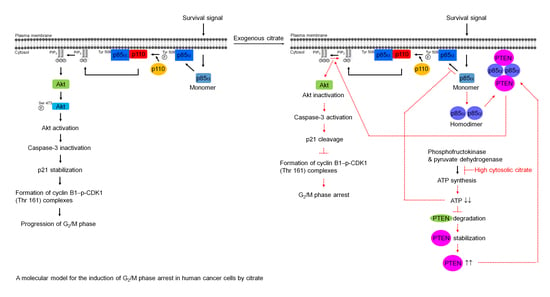
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
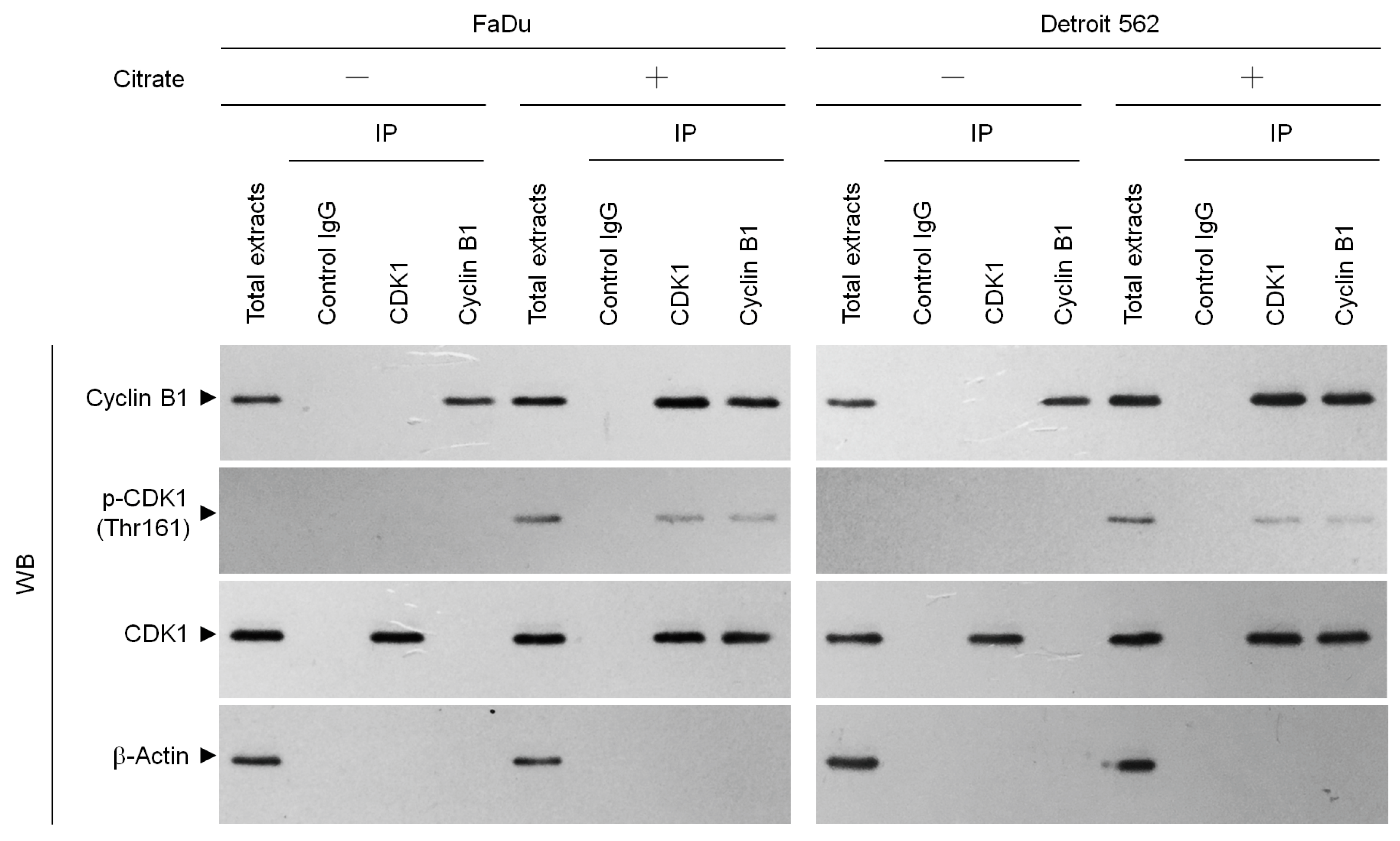
2.1. Citrate Induces G2/M-Phase Arrest in Human Cancer Cells by Stabilizing the Formation of Cyclin B1–p-CDK1 (Thr 161) Complexes
2.2. Suppression of Akt-Mediated Signaling is Required for Citrate-Induced G2/M Phase Arrest
2.3. p85α–PTEN Complex-Mediated Inhibition of Akt is Required for Citrate-Induced G2/M Phase Arrest
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemicals, Reagents, and Plasmids
4.3. Antibodies
4.4. Cell Viability Assay
4.5. Cell Proliferation Assay
4.6. Measurement of the Cell Cycle by Flow Cytometry
4.7. Western Blot Analysis
4.8. Plasmid Transfection
4.9. Subcellular Fractionation
4.10. Coimmunoprecipitation
4.11. Statistical Analysis of Data
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACC | Acetyl-CoA carboxylase |
| ACL | ATP-citrate lyase |
| CDK1 | Cyclin-dependent kinase 1 |
| OAA | Oxaloacetate |
| PTEN | Phosphatase and tensin homolog deleted from chromosome 10 |
| PI3K | Phosphatidylinositol 3-kinase |
| PIP2 | Phosphatidylinositol-4,5-bisphosphate |
| PIP3 | Phosphatidylinositol-3,4,5-trisphosphate |
| Akt | Protein kinase B |
| PSC | Human pharyngeal squamous carcinoma |
References
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef]
- Dang, C.V.; Semenza, G.L. Oncogenic alterations of metabolism. Trends Biochem. Sci. 1999, 24, 68–72. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-citrate lyase: A key player in cancer metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Lee, M.Y.; Park, S.W.; Moon, J.S.; Koh, Y.K.; Ahn, Y.H.; Park, B.W.; Kim, K.S. Up-regulation of acetyl-CoA carboxylase alpha and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. J. Biol. Chem. 2007, 282, 26122–26131. [Google Scholar] [CrossRef]
- Chajes, V.; Cambot, M.; Moreau, K.; Lenoir, G.M.; Joulin, V. Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer Res. 2006, 66, 5287–5294. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; Vanderhoydonc, F.; Elgamal, A.A.; Eelen, M.; Vercaeren, I.; Joniau, S.; Van Poppel, H.; Baert, L.; Goossens, K.; Heyns, W.; Verhoeven, G. Selective activation of the fatty acid synthesis pathway in human prostate cancer. Int. J. Cancer 2000, 88, 176–179. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, C.M.; Yang, J.; Sims, H.F.; Gross, R.W. Reversible high affinity inhibition of phosphofructokinase-1 by acyl-CoA: A mechanism integrating glycolytic flux with lipid metabolism. J. Biol. Chem. 2011, 286, 11937–11950. [Google Scholar] [CrossRef]
- Taylor, W.M.; Halperin, M.L. Regulation of pyruvate dehydrogenase in muscle. Inhibition by citrate. J. Biol. Chem. 1973, 248, 6080–6083. [Google Scholar]
- Halperin, M.L.; Cheema-Dhadli, S.; Taylor, W.M.; Fritz, I.B. Role of the citrate transporter in the control of fatty acid synthesis. Adv. Enzyme Regul. 1975, 13, 435–445. [Google Scholar] [CrossRef]
- Xu, R.H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [Google Scholar]
- Zhou, Y.; Tozzi, F.; Chen, J.; Fan, F.; Xia, L.; Wang, J.; Gao, G.; Zhang, A.; Xia, X.; Brasher, H.; Widger, W.; Ellis, L.M.; Weihua, Z. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res. 2012, 72, 304–314. [Google Scholar] [CrossRef]
- Brazil, D.P.; Yang, Z.Z.; Hemmings, B.A. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem. Sci. 2004, 29, 233–242. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Chagpar, R.B.; Links, P.H.; Pastor, M.C.; Furber, L.A.; Hawrysh, A.D.; Chamberlain, M.D.; Anderson, D.H. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2010, 107, 5471–5476. [Google Scholar] [CrossRef] [Green Version]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef]
- Franke, T.F.; Hornik, C.P.; Segev, L.; Shostak, G.A.; Sugimoto, C. PI3K/Akt and apoptosis: Size matters. Oncogene 2003, 22, 8983–8998. [Google Scholar] [CrossRef]
- Katayama, K.; Fujita, N.; Tsuruo, T. Akt/protein kinase B-dependent phosphorylation and inactivation of WEE1Hu promote cell cycle progression at G2/M transition. Mol. Cell. Biol. 2005, 25, 5725–5737. [Google Scholar] [CrossRef]
- Li, Y.; Dowbenko, D.; Lasky, L.A. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J. Biol. Chem. 2002, 277, 11352–11361. [Google Scholar] [CrossRef]
- Li, W.; Fan, J.; Banerjee, D.; Bertino, J.R. Overexpression of p21(waf1) decreases G2-M arrest and apoptosis induced by paclitaxel in human sarcoma cells lacking both p53 and functional Rb protein. Mol. Pharmacol. 1999, 55, 1088–1093. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Fox, C.J.; Plas, D.R.; Hammerman, P.S.; Cinalli, R.M.; Thompson, C.B. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell. Biol. 2003, 23, 7315–7328. [Google Scholar] [CrossRef]
- Bauer, D.E.; Hatzivassiliou, G.; Zhao, F.; Andreadis, C.; Thompson, C.B. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 2005, 24, 6314–6322. [Google Scholar] [CrossRef] [Green Version]
- Buzzai, M.; Bauer, D.E.; Jones, R.G.; Deberardinis, R.J.; Hatzivassiliou, G.; Elstrom, R.L.; Thompson, C.B. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene 2005, 24, 4165–4173. [Google Scholar] [CrossRef]
- Berwick, D.C.; Hers, I.; Heesom, K.J.; Moule, S.K.; Tavare, J.M. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J. Biol. Chem. 2002, 277, 33895–33900. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; Thompson, C.B. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef]
- Icard, P.; Poulain, L.; Lincet, H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Biophys. Acta 2012, 1825, 111–116. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, X.; Zhang, H.; Lan, J.; Huang, G.; Varin, E.; Lincet, H.; Poulain, L.; Icard, P. Citrate induces apoptotic cell death: A promising way to treat gastric carcinoma? Anticancer Res. 2011, 31, 797–805. [Google Scholar]
- Lincet, H.; Kafara, P.; Giffard, F.; Abeilard-Lemoisson, E.; Duval, M.; Louis, M.H.; Poulain, L.; Icard, P. Inhibition of Mcl-1 expression by citrate enhances the effect of Bcl-xL inhibitors on human ovarian carcinoma cells. J. Ovarian Res. 2013, 6, 72. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, R.; Yu, Z.; Li, R.; Li, J.; Zhao, X.; Song, S.; Liu, J.; Huang, G. Dichloroacetate restores drug sensitivity in paclitaxel-resistant cells by inducing citric acid accumulation. Mol. Cancer 2015, 14, 63. [Google Scholar] [CrossRef]
- Guimaraes, L.F.; Fidalgo, T.K.; Menezes, G.C.; Primo, L.G.; Costa e Silva-Filho, F. Effects of citric acid on cultured human osteoblastic cells. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2010, 110, 665–669. [Google Scholar] [CrossRef]
- Nurse, P. Universal control mechanism regulating onset of M-phase. Nature 1990, 344, 503–508. [Google Scholar] [CrossRef]
- Pines, J.; Hunter, T. Isolation of a human cyclin cDNA: Evidence for cyclin mRNA and protein regulation in the cell cycle and for interaction with p34cdc2. Cell 1989, 58, 833–846. [Google Scholar] [CrossRef]
- Fung, T.K.; Poon, R.Y. A roller coaster ride with the mitotic cyclins. Semin. Cell. Dev. Biol. 2005, 16, 335–342. [Google Scholar] [CrossRef]
- Charrier-Savournin, F.B.; Chateau, M.T.; Gire, V.; Sedivy, J.; Piette, J.; Dulic, V. p21-Mediated nuclear retention of cyclin B1-Cdk1 in response to genotoxic stress. Molecular Biol. Cell 2004, 15, 3965–3976. [Google Scholar] [CrossRef]
- Jin, Y.H.; Yoo, K.J.; Lee, Y.H.; Lee, S.K. Caspase 3-mediated cleavage of p21WAF1/CIP1 associated with the cyclin A-cyclin-dependent kinase 2 complex is a prerequisite for apoptosis in SK-HEP-1 cells. J. Biol. Chem. 2000, 275, 30256–30263. [Google Scholar] [CrossRef]
- Ross, A.H.; Gericke, A. Phosphorylation keeps PTEN phosphatase closed for business. Proc. Natl. Acad. Sci. USA 2009, 106, 1297–1298. [Google Scholar] [CrossRef] [Green Version]
- Cheung, L.W.; Walkiewicz, K.W.; Besong, T.M.; Guo, H.; Hawke, D.H.; Arold, S.T.; Mills, G.B. Regulation of the PI3K pathway through a p85alpha monomer-homodimer equilibrium. eLife 2015, 4, e06866. [Google Scholar] [CrossRef]
- Jang, M.; Kim, S.S.; Lee, J. Cancer cell metabolism: Implications for therapeutic targets. Exp. Mol. Med. 2013, 45, e45. [Google Scholar] [CrossRef]
- Kavanaugh, W.M.; Turck, C.W.; Klippel, A.; Williams, L.T. Tyrosine 508 of the 85-kilodalton subunit of phosphatidylinositol 3-kinase is phosphorylated by the platelet-derived growth factor receptor. Biochemistry 1994, 33, 11046–11050. [Google Scholar] [CrossRef]
- Skolnik, E.Y.; Margolis, B.; Mohammadi, M.; Lowenstein, E.; Fischer, R.; Drepps, A.; Ullrich, A.; Schlessinger, J. Cloning of PI3 kinase-associated p85 utilizing a novel method for expression/cloning of target proteins for receptor tyrosine kinases. Cell 1991, 65, 83–90. [Google Scholar] [CrossRef]
- Nolte, R.T.; Eck, M.J.; Schlessinger, J.; Shoelson, S.E.; Harrison, S.C. Crystal structure of the PI 3-kinase p85 amino-terminal SH2 domain and its phosphopeptide complexes. Nat. Struct. Biol. 1996, 3, 364–374. [Google Scholar] [CrossRef]
- Hiles, I.D.; Otsu, M.; Volinia, S.; Fry, M.J.; Gout, I.; Dhand, R.; Panayotou, G.; Ruiz-Larrea, F.; Thompson, A.; Totty, N.F.; et al. Phosphatidylinositol 3-kinase: Structure and expression of the 110 kd catalytic subunit. Cell 1992, 70, 419–429. [Google Scholar] [CrossRef]
- Klippel, A.; Escobedo, J.A.; Hirano, M.; Williams, L.T. The interaction of small domains between the subunits of phosphatidylinositol 3-kinase determines enzyme activity. Mol. Cell. Biol. 1994, 14, 2675–2685. [Google Scholar] [CrossRef]
- Schramp, M.; Hedman, A.; Li, W.; Tan, X.; Anderson, R. PIP kinases from the cell membrane to the nucleus. Sub-Cell. Biochem. 2012, 58, 25–59. [Google Scholar]
- Maddika, S.; Kavela, S.; Rani, N.; Palicharla, V.R.; Pokorny, J.L.; Sarkaria, J.N.; Chen, J. WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 2011, 13, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Smits, V.A.; Klompmaker, R.; Vallenius, T.; Rijksen, G.; Makela, T.P.; Medema, R.H. p21 inhibits Thr161 phosphorylation of Cdc2 to enforce the G2 DNA damage checkpoint. J. Biol. Chem. 2000, 275, 30638–30643. [Google Scholar] [CrossRef]
- Dhand, R.; Hiles, I.; Panayotou, G.; Roche, S.; Fry, M.J.; Gout, I.; Totty, N.F.; Truong, O.; Vicendo, P.; Yonezawa, K.; et al. PI 3-kinase is a dual specificity enzyme: Autoregulation by an intrinsic protein-serine kinase activity. EMBO J. 1994, 13, 522–533. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, Y.; McIlroy, J.; Rordorf-Nikolic, T.; Orr, G.A.; Backer, J.M. Regulation of the p85/p110 phosphatidylinositol 3’-kinase: Stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol. Cell. Biol. 1998, 18, 1379–1387. [Google Scholar] [CrossRef]
- Foukas, L.C.; Beeton, C.A.; Jensen, J.; Phillips, W.A.; Shepherd, P.R. Regulation of phosphoinositide 3-kinase by its intrinsic serine kinase activity in vivo. Mol. Cell. Biol. 2004, 24, 966–975. [Google Scholar] [CrossRef]
- Ren, J.G.; Seth, P.; Ye, H.; Guo, K.; Hanai, J.I.; Husain, Z.; Sukhatme, V.P. Citrate Suppresses Tumor Growth in Multiple Models through Inhibition of Glycolysis, the Tricarboxylic Acid Cycle and the IGF-1R Pathway. Sci. Rep. 2017, 7, 4537. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.L.; Chen, S.S.; Huang, R.Y.; Lu, Y.C.; Liao, Y.R.; Reddy, M.V.; Lee, C.C.; Wu, T.S. Suppression of PI3K/Akt signaling by synthetic bichalcone analog TSWU-CD4 induces ER stress- and Bax/Bak-mediated apoptosis of cancer cells. Apoptosis 2014, 19, 1637–1653. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.L.; Lu, Y.C.; Chung, J.G.; Wang, S.G.; Lin, H.T.; Kang, S.E.; Tang, C.H.; Ko, J.L.; Chen, S.S. Down-regulation of MMP-2 through the p38 MAPK-NF-kappaB-dependent pathway by aloe-emodin leads to inhibition of nasopharyngeal carcinoma cell invasion. Mol. Carcinog. 2010, 49, 783–797. [Google Scholar] [PubMed]
- Taha, M.S.; Nouri, K.; Milroy, L.G.; Moll, J.M.; Herrmann, C.; Brunsveld, L.; Piekorz, R.P.; Ahmadian, M.R. Subcellular fractionation and localization studies reveal a direct interaction of the fragile X mental retardation protein (FMRP) with nucleolin. PLoS ONE 2014, 9, e91465. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, K.-C.; Wang, S.-G.; Lin, M.-L.; Chen, S.-S. Citrate-Induced p85α–PTEN Complex Formation Causes G2/M Phase Arrest in Human Pharyngeal Squamous Carcinoma Cell Lines. Int. J. Mol. Sci. 2019, 20, 2105. https://doi.org/10.3390/ijms20092105
Hung K-C, Wang S-G, Lin M-L, Chen S-S. Citrate-Induced p85α–PTEN Complex Formation Causes G2/M Phase Arrest in Human Pharyngeal Squamous Carcinoma Cell Lines. International Journal of Molecular Sciences. 2019; 20(9):2105. https://doi.org/10.3390/ijms20092105
Chicago/Turabian StyleHung, Kuang-Chen, Shyang-Guang Wang, Meng-Liang Lin, and Shih-Shun Chen. 2019. "Citrate-Induced p85α–PTEN Complex Formation Causes G2/M Phase Arrest in Human Pharyngeal Squamous Carcinoma Cell Lines" International Journal of Molecular Sciences 20, no. 9: 2105. https://doi.org/10.3390/ijms20092105
APA StyleHung, K. -C., Wang, S. -G., Lin, M. -L., & Chen, S. -S. (2019). Citrate-Induced p85α–PTEN Complex Formation Causes G2/M Phase Arrest in Human Pharyngeal Squamous Carcinoma Cell Lines. International Journal of Molecular Sciences, 20(9), 2105. https://doi.org/10.3390/ijms20092105