Role of Cannabinoid Receptor Type 1 in Insulin Resistance and Its Biological Implications


Abstract
:1. Introduction
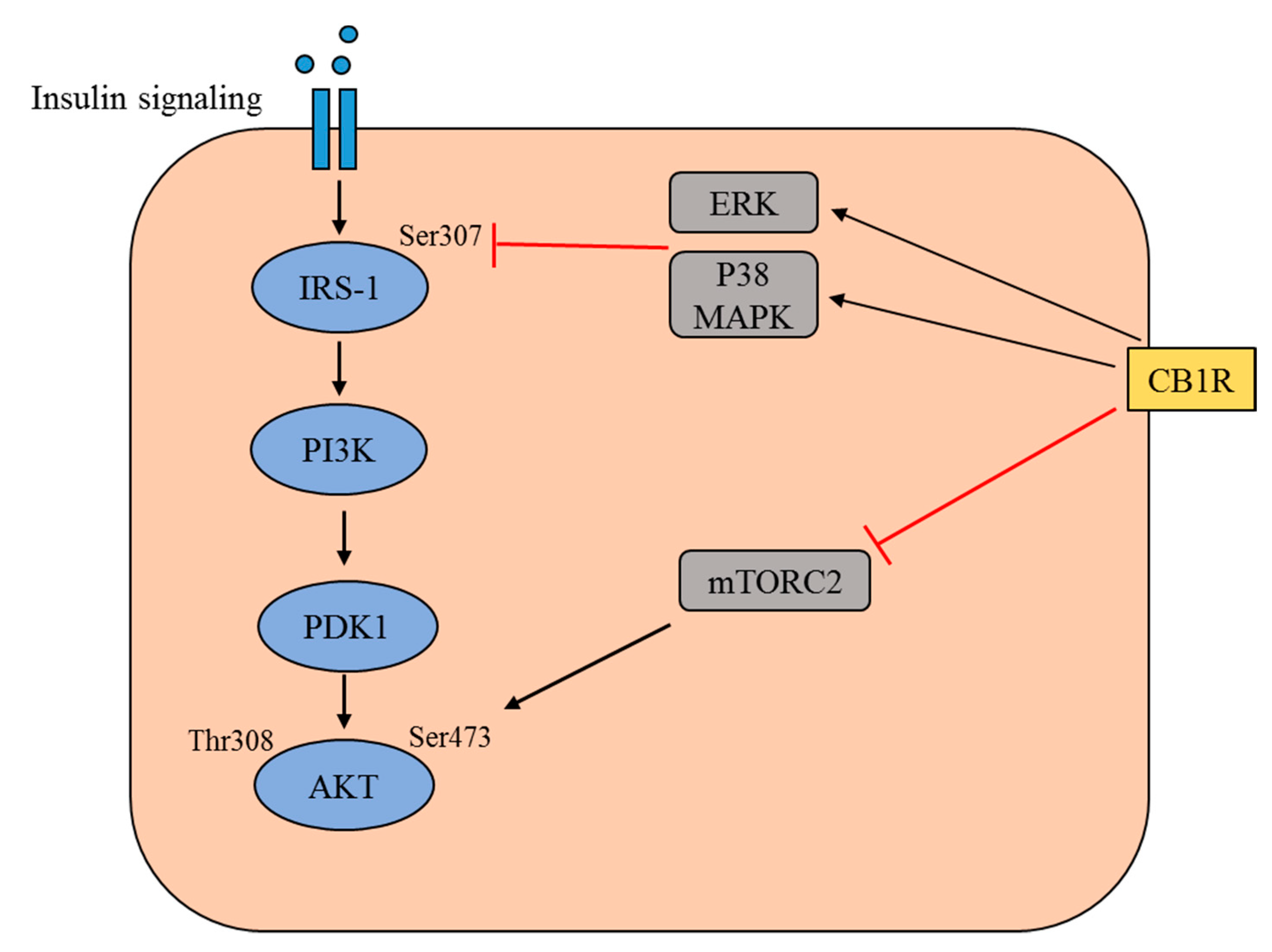
2. Insulin Signaling Pathways
3. Insulin Resistance
4. Distribution and Biological Roles of CB1R in the Human Body
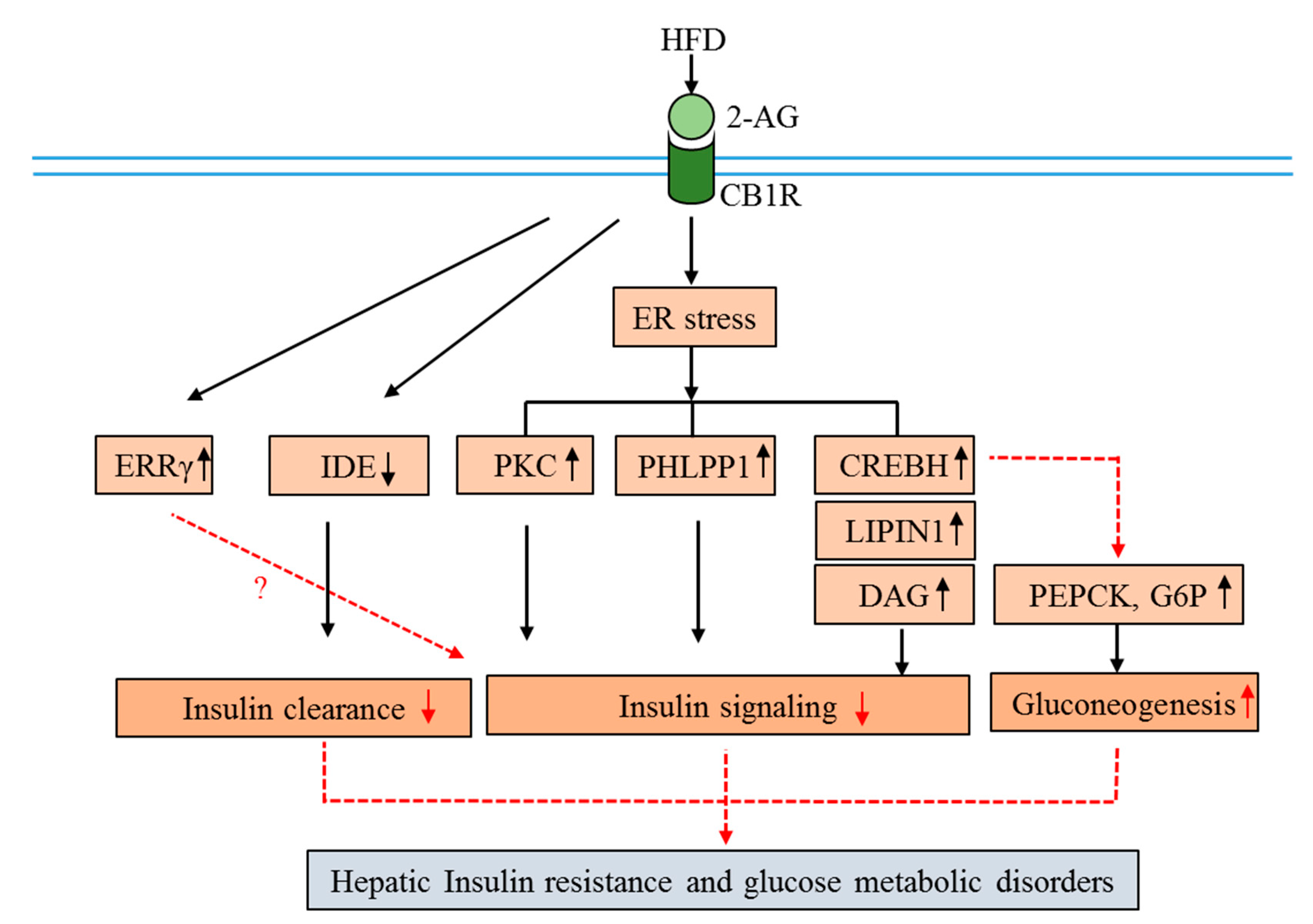
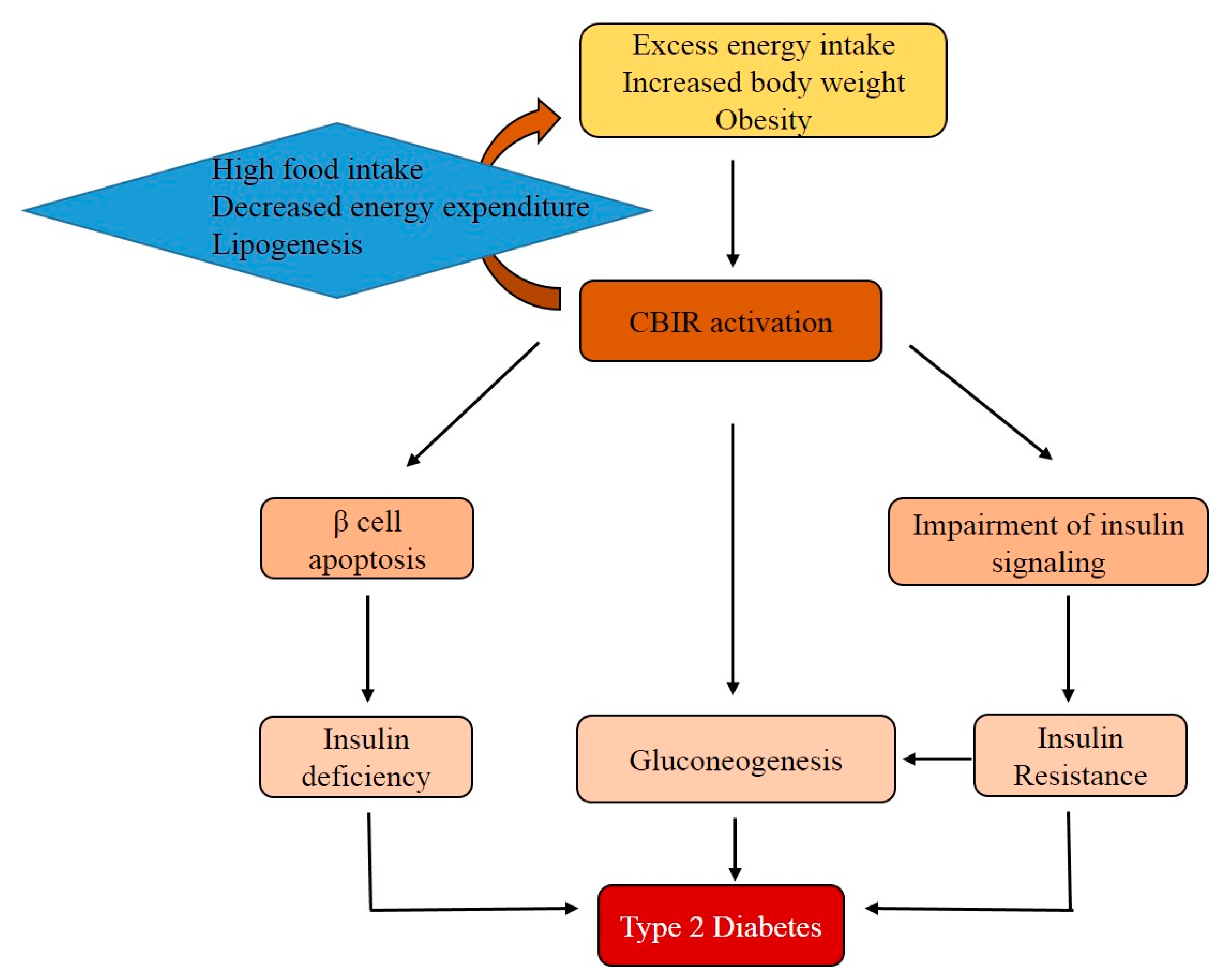
5. Participation of CB1R in Obesity and Insulin Resistance
6. The Functions of CB1R in Pancreatic β-Cells and Skeletal Muscle
7. The Therapeutic Potential
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Finck, B.N. Targeting metabolism, insulin resistance, and diabetes to treat nonalcoholic steatohepatitis. Diabetes 2018, 67, 2485–2493. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacher, P.; Batkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed]
- Simon, V.; Cota, D. Mechanisms in endocrinology: Endocannabinoids and metabolism: Past, present and future. Eur. J. Endocrinol. 2017, 176, R309–R324. [Google Scholar] [CrossRef] [PubMed]
- Matias, I.; Di Marzo, V. Endocannabinoids and the control of energy balance. Trends Endocrinol. Metab. 2007, 18, 27–37. [Google Scholar] [CrossRef]
- Kunos, G. Understanding metabolic homeostasis and imbalance: What is the role of the endocannabinoid system? Am. J. Med. 2007, 120, S18–S24. [Google Scholar] [CrossRef]
- Mazier, W.; Saucisse, N.; Gatta-Cherifi, B.; Cota, D. The endocannabinoid system: Pivotal orchestrator of obesity and metabolic disease. Trends Endocrinol. Metab. 2015, 26, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, K.; Sell, H.; Taube, A.; Koenen, M.; Platzbecker, B.; Cramer, A.; Horrighs, A.; Lehtonen, M.; Tennagels, N.; Eckel, J. Cannabinoid type 1 receptors in human skeletal muscle cells participate in the negative crosstalk between fat and muscle. Diabetologia 2009, 52, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, L.; Xiong, K.; Godlewski, G.; Mukhopadhyay, B.; Tam, J.; Yin, S.; Gao, P.; Shan, X.; Pickel, J.; et al. Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology 2012, 142, 1218–1228 e1211. [Google Scholar] [CrossRef] [PubMed]
- Motaghedi, R.; McGraw, T.E. The cb1 endocannabinoid system modulates adipocyte insulin sensitivity. Obesity 2008, 16, 1727–1734. [Google Scholar] [CrossRef]
- Jbilo, O.; Ravinet-Trillou, C.; Arnone, M.; Buisson, I.; Bribes, E.; Peleraux, A.; Penarier, G.; Soubrie, P.; Le Fur, G.; Galiegue, S.; et al. The cb1 receptor antagonist rimonabant reverses the diet-induced obesity phenotype through the regulation of lipolysis and energy balance. FASEB J. 2005, 19, 1567–1569. [Google Scholar] [CrossRef]
- Engeli, S.; Bohnke, J.; Feldpausch, M.; Gorzelniak, K.; Janke, J.; Batkai, S.; Pacher, P.; Harvey-White, J.; Luft, F.C.; Sharma, A.M.; et al. Activation of the peripheral endocannabinoid system in human obesity. Diabetes 2005, 54, 2838–2843. [Google Scholar] [CrossRef] [PubMed]
- Cota, D.; Marsicano, G.; Tschop, M.; Grubler, Y.; Flachskamm, C.; Schubert, M.; Auer, D.; Yassouridis, A.; Thone-Reineke, C.; Ortmann, S.; et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J. Clin. Investig. 2003, 112, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pi-Sunyer, F.X.; Aronne, L.J.; Heshmati, H.M.; Devin, J.; Rosenstock, J.; Group, R.I.-N.A.S. Effect of rimonabant, a cannabinoid-1 receptor blocker, on weight and cardiometabolic risk factors in overweight or obese patients: Rio-north america: A randomized controlled trial. JAMA 2006, 295, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Faerch, K.; Vistisen, D.; Pacini, G.; Torekov, S.S.; Johansen, N.B.; Witte, D.R.; Jonsson, A.; Pedersen, O.; Hansen, T.; Lauritzen, T.; et al. Insulin resistance is accompanied by increased fasting glucagon and delayed glucagon suppression in individuals with normal and impaired glucose regulation. Diabetes 2016, 65, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Bevan, P. Insulin signalling. J. Cell Sci. 2001, 114, 1429–1430. [Google Scholar] [PubMed]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins irs1 and irs2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef]
- Ho, C.K.; Sriram, G.; Dipple, K.M. Insulin sensitivity predictions in individuals with obesity and type ii diabetes mellitus using mathematical model of the insulin signal transduction pathway. Mol. Genet. Metab. 2016, 119, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Yip, S.C.; Saha, S.; Chernoff, J. Ptp1b: A double agent in metabolism and oncogenesis. Trends Biochem. Sci. 2010, 35, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Fantus, I.G. Inhibition of the protein tyrosine phosphatase ptp1b: Potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 621–640. [Google Scholar] [CrossRef] [PubMed]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.C.; et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1b gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, F.; Ge, X.; Yan, T.; Chen, X.; Shi, X.; Zhai, Q. Sirt1 improves insulin sensitivity under insulin-resistant conditions by repressing ptp1b. Cell Metab. 2007, 6, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Klaman, L.D.; Boss, O.; Peroni, O.D.; Kim, J.K.; Martino, J.L.; Zabolotny, J.M.; Moghal, N.; Lubkin, M.; Kim, Y.B.; Sharpe, A.H.; et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1b-deficient mice. Mol. Cell Biol. 2000, 20, 5479–5489. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Aljada, A.; Bandyopadhyay, A. Inflammation: The link between insulin resistance, obesity and diabetes. Trends Immunol. 2004, 25, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Peraldi, P.; Spiegelman, B. Tnf-alpha and insulin resistance: Summary and future prospects. Mol. Cell Biochem. 1998, 182, 169–175. [Google Scholar] [CrossRef]
- de Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Hundal, R.S.; Petersen, K.F.; Mayerson, A.B.; Randhawa, P.S.; Inzucchi, S.; Shoelson, S.E.; Shulman, G.I. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J. Clin. Investig. 2002, 109, 1321–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Maddux, B.A.; Goldfine, I.D. The molecular basis for oxidative stress-induced insulin resistance. Antioxid. Redox Signal. 2005, 7, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Mohammadi, M.T.; Rezaee, R.; Sahebkar, A. Crocin improves renal function by declining nox-4, il-18, and p53 expression levels in an experimental model of diabetic nephropathy. J. Cell Biochem. 2018, 119, 6080–6093. [Google Scholar] [CrossRef] [PubMed]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Avruch, J. Insulin signal transduction through protein kinase cascades. Mol. Cell Biochem. 1998, 182, 31–48. [Google Scholar] [CrossRef]
- Beeson, M.; Sajan, M.P.; Dizon, M.; Grebenev, D.; Gomez-Daspet, J.; Miura, A.; Kanoh, Y.; Powe, J.; Bandyopadhyay, G.; Standaert, M.L.; et al. Activation of protein kinase c-zeta by insulin and phosphatidylinositol-3,4,5-(po4)3 is defective in muscle in type 2 diabetes and impaired glucose tolerance: Amelioration by rosiglitazone and exercise. Diabetes 2003, 52, 1926–1934. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for jnk in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Sesti, G. Pathophysiology of insulin resistance. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 665–679. [Google Scholar] [CrossRef]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes er stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef]
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Yoshiuchi, K.; Hatazaki, M.; Matsuoka, T.A.; Ozawa, K.; Ogawa, S.; Hori, M.; Yamasaki, Y.; et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J. Biol. Chem. 2005, 280, 847–851. [Google Scholar] [CrossRef]
- Mackie, K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb Exp. Pharmacol. 2005, 299–325. [Google Scholar] [CrossRef]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef]
- Di Marzo, V.; Stella, N.; Zimmer, A. Endocannabinoid signalling and the deteriorating brain. Nat. Rev. Neurosci. 2015, 16, 30–42. [Google Scholar] [CrossRef]
- Iversen, L. Cannabis and the brain. Brain 2003, 126, 1252–1270. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.M.; Kirkham, T.C. Anandamide induces overeating: Mediation by central cannabinoid (cb1) receptors. Psychopharmacology 1999, 143, 315–317. [Google Scholar] [CrossRef]
- Colombo, G.; Agabio, R.; Diaz, G.; Lobina, C.; Reali, R.; Gessa, G.L. Appetite suppression and weight loss after the cannabinoid antagonist sr 141716. Life Sci. 1998, 63, PL113–PL117. [Google Scholar] [CrossRef]
- Werner, N.A.; Koch, J.E. Effects of the cannabinoid antagonists am281 and am630 on deprivation-induced intake in lewis rats. Brain Res. 2003, 967, 290–292. [Google Scholar] [CrossRef]
- Croci, T.; Manara, L.; Aureggi, G.; Guagnini, F.; Rinaldi-Carmona, M.; Maffrand, J.P.; Le Fur, G.; Mukenge, S.; Ferla, G. In vitro functional evidence of neuronal cannabinoid cb1 receptors in human ileum. Br. J. Pharmacol. 1998, 125, 1393–1395. [Google Scholar] [CrossRef]
- Jamshidi, N.; Taylor, D.A. Anandamide administration into the ventromedial hypothalamus stimulates appetite in rats. Br. J. Pharmacol. 2001, 134, 1151–1154. [Google Scholar] [CrossRef] [Green Version]
- Riedel, G.; Davies, S.N. Cannabinoid function in learning, memory and plasticity. Handb Exp. Pharmacol. 2005, 445–477. [Google Scholar] [CrossRef]
- Ramirez, B.G.; Blazquez, C.; Gomez del Pulgar, T.; Guzman, M.; de Ceballos, M.L. Prevention of alzheimer’s disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Molina-Holgado, F.; Pinteaux, E.; Moore, J.D.; Molina-Holgado, E.; Guaza, C.; Gibson, R.M.; Rothwell, N.J. Endogenous interleukin-1 receptor antagonist mediates anti-inflammatory and neuroprotective actions of cannabinoids in neurons and glia. J. Neurosci. 2003, 23, 6470–6474. [Google Scholar] [CrossRef]
- Maccarrone, M.; Bab, I.; Biro, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after thc. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Trembovler, V.; Di Marzo, V.; Petrosino, S.; Leo, G.; Alexandrovich, A.; Regev, E.; Casap, N.; Shteyer, A.; Ledent, C.; et al. The cannabinoid cb1 receptor regulates bone formation by modulating adrenergic signaling. FASEB J. 2008, 22, 285–294. [Google Scholar] [CrossRef]
- Clapper, J.R.; Moreno-Sanz, G.; Russo, R.; Guijarro, A.; Vacondio, F.; Duranti, A.; Tontini, A.; Sanchini, S.; Sciolino, N.R.; Spradley, J.M.; et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat. Neurosci. 2010, 13, 1265–1270. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.K.; Devi, L.A. The highs and lows of cannabinoid receptor expression in disease: Mechanisms and their therapeutic implications. Pharmacol. Rev. 2011, 63, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, R. Type i cannabinoid receptor trafficking: All roads lead to lysosome. Traffic 2011, 12, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Di Marzo, V. At the heart of the matter: The endocannabinoid system in cardiovascular function and dysfunction. Trends Pharmacol. Sci. 2012, 33, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Sterin-Borda, L.; Del Zar, C.F.; Borda, E. Differential cb1 and cb2 cannabinoid receptor-inotropic response of rat isolated atria: Endogenous signal transduction pathways. Biochem. Pharmacol. 2005, 69, 1705–1713. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Batkai, S.; Patel, V.; Kashiwaya, Y.; Liaudet, L.; Evgenov, O.V.; Mackie, K.; Hasko, G.; Pacher, P. Cb1 cannabinoid receptors promote oxidative stress and cell death in murine models of doxorubicin-induced cardiomyopathy and in human cardiomyocytes. Cardiovasc. Res. 2010, 85, 773–784. [Google Scholar] [CrossRef]
- Rajesh, M.; Batkai, S.; Kechrid, M.; Mukhopadhyay, P.; Lee, W.S.; Horvath, B.; Holovac, E.; Cinar, R.; Liaudet, L.; Mackie, K.; et al. Cannabinoid 1 receptor promotes cardiac dysfunction, oxidative stress, inflammation, and fibrosis in diabetic cardiomyopathy. Diabetes 2012, 61, 716–727. [Google Scholar] [CrossRef]
- Piotrowska, Z.; Niezgoda, M.; Lebkowski, W.; Filipek, A.; Domian, N.; Kasacka, I. Sex differences in distribution of cannabinoid receptors (cb1 and cb2), s100a6 and cacybp/sip in human ageing hearts. Biol. Sex. Differ. 2018, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Slavic, S.; Lauer, D.; Sommerfeld, M.; Kemnitz, U.R.; Grzesiak, A.; Trappiel, M.; Thone-Reineke, C.; Baulmann, J.; Paulis, L.; Kappert, K.; et al. Cannabinoid receptor 1 inhibition improves cardiac function and remodelling after myocardial infarction and in experimental metabolic syndrome. J. Mol. Med. 2013, 91, 811–823. [Google Scholar] [CrossRef]
- Lim, S.Y.; Davidson, S.M.; Yellon, D.M.; Smith, C.C. The cannabinoid cb1 receptor antagonist, rimonabant, protects against acute myocardial infarction. Basic Res. Cardiol. 2009, 104, 781–792. [Google Scholar] [CrossRef]
- Lopez-Cardona, A.P.; Perez-Cerezales, S.; Fernandez-Gonzalez, R.; Laguna-Barraza, R.; Pericuesta, E.; Agirregoitia, N.; Gutierrez-Adan, A.; Agirregoitia, E. Cb1 cannabinoid receptor drives oocyte maturation and embryo development via pi3k/akt and mapk pathways. FASEB J. 2017, 31, 3372–3382. [Google Scholar] [CrossRef] [PubMed]
- Agirregoitia, E.; Totorikaguena, L.; Exposito, A.; Mendoza, R.; Matorras, R.; Agirregoitia, N. Dynamic of expression and localization of cannabinoid-degrading enzymes faah and mgll in relation to cb1 during meiotic maturation of human oocytes. Cell Tissue Res. 2016, 365, 393–401. [Google Scholar] [CrossRef]
- Gebeh, A.K.; Willets, J.M.; Marczylo, E.L.; Taylor, A.H.; Konje, J.C. Ectopic pregnancy is associated with high anandamide levels and aberrant expression of faah and cb1 in fallopian tubes. J. Clin. Endocrinol. Metab. 2012, 97, 2827–2835. [Google Scholar] [CrossRef]
- Wolfson, M.L.; Correa, F.; Leishman, E.; Vercelli, C.; Cymeryng, C.; Blanco, J.; Bradshaw, H.B.; Franchi, A.M. Lipopolysaccharide-induced murine embryonic resorption involves changes in endocannabinoid profiling and alters progesterone secretion and inflammatory response by a cb1-mediated fashion. Mol. Cell Endocrinol. 2015, 411, 214–222. [Google Scholar] [CrossRef]
- Izzo, A.A.; Sharkey, K.A. Cannabinoids and the gut: New developments and emerging concepts. Pharmacol. Ther. 2010, 126, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, K.A.; Wiley, J.W. The role of the endocannabinoid system in the brain-gut axis. Gastroenterology 2016, 151, 252–266. [Google Scholar] [CrossRef] [Green Version]
- Keenan, C.M.; Storr, M.A.; Thakur, G.A.; Wood, J.T.; Wager-Miller, J.; Straiker, A.; Eno, M.R.; Nikas, S.P.; Bashashati, M.; Hu, H.; et al. Am841, a covalent cannabinoid ligand, powerfully slows gastrointestinal motility in normal and stressed mice in a peripherally restricted manner. Br. J. Pharmacol. 2015, 172, 2406–2418. [Google Scholar] [CrossRef] [PubMed]
- Pagotto, U.; Marsicano, G.; Cota, D.; Lutz, B.; Pasquali, R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr. Rev. 2006, 27, 73–100. [Google Scholar] [CrossRef] [PubMed]
- Matias, I.; Gonthier, M.P.; Orlando, P.; Martiadis, V.; De Petrocellis, L.; Cervino, C.; Petrosino, S.; Hoareau, L.; Festy, F.; Pasquali, R.; et al. Regulation, function, and dysregulation of endocannabinoids in models of adipose and beta-pancreatic cells and in obesity and hyperglycemia. J. Clin. Endocrinol. Metab. 2006, 91, 3171–3180. [Google Scholar] [CrossRef]
- Matias, I.; Bisogno, T.; Di Marzo, V. Endogenous cannabinoids in the brain and peripheral tissues: Regulation of their levels and control of food intake. Int. J. Obes. 2006, 30, S7–S12. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, L.; Valerio, A.; Dossena, M.; Cardile, A.; Ragni, M.; Pagano, C.; Pagotto, U.; Carruba, M.O.; Vettor, R.; Nisoli, E. Cannabinoid receptor stimulation impairs mitochondrial biogenesis in mouse white adipose tissue, muscle, and liver: The role of enos, p38 mapk, and ampk pathways. Diabetes 2010, 59, 2826–2836. [Google Scholar] [CrossRef] [PubMed]
- Perwitz, N.; Fasshauer, M.; Klein, J. Cannabinoid receptor signaling directly inhibits thermogenesis and alters expression of adiponectin and visfatin. Horm. Metab. Res. 2006, 38, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Ravinet Trillou, C.; Delgorge, C.; Menet, C.; Arnone, M.; Soubrie, P. Cb1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int. J. Obes. Relat. Metab. Disord 2004, 28, 640–648. [Google Scholar] [CrossRef]
- Ge, Q.; Maury, E.; Rycken, L.; Gerard, J.; Noel, L.; Detry, R.; Navez, B.; Brichard, S.M. Endocannabinoids regulate adipokine production and the immune balance of omental adipose tissue in human obesity. Int. J. Obes. 2013, 37, 874–880. [Google Scholar] [CrossRef]
- Lipina, C.; Stretton, C.; Hastings, S.; Hundal, J.S.; Mackie, K.; Irving, A.J.; Hundal, H.S. Regulation of map kinase-directed mitogenic and protein kinase b-mediated signaling by cannabinoid receptor type 1 in skeletal muscle cells. Diabetes 2010, 59, 375–385. [Google Scholar] [CrossRef]
- Arrabal, S.; Lucena, M.A.; Canduela, M.J.; Ramos-Uriarte, A.; Rivera, P.; Serrano, A.; Pavon, F.J.; Decara, J.; Vargas, A.; Baixeras, E.; et al. Pharmacological blockade of cannabinoid cb1 receptors in diet-induced obesity regulates mitochondrial dihydrolipoamide dehydrogenase in muscle. PLoS ONE 2015, 10, e0145244. [Google Scholar] [CrossRef]
- Iannotti, F.A.; Silvestri, C.; Mazzarella, E.; Martella, A.; Calvigioni, D.; Piscitelli, F.; Ambrosino, P.; Petrosino, S.; Czifra, G.; Biro, T.; et al. The endocannabinoid 2-ag controls skeletal muscle cell differentiation via cb1 receptor-dependent inhibition of kv7 channels. Proc. Natl. Acad. Sci. USA 2014, 111, E2472–E2481. [Google Scholar] [CrossRef]
- Mendizabal-Zubiaga, J.; Melser, S.; Benard, G.; Ramos, A.; Reguero, L.; Arrabal, S.; Elezgarai, I.; Gerrikagoitia, I.; Suarez, J.; Rodriguez De Fonseca, F.; et al. Cannabinoid cb1 receptors are localized in striated muscle mitochondria and regulate mitochondrial respiration. Front. Physiol. 2016, 7, 476. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Mariscal, I.; Montoro, R.A.; O’Connell, J.F.; Kim, Y.; Gonzalez-Freire, M.; Liu, Q.R.; Alfaras, I.; Carlson, O.D.; Lehrmann, E.; Zhang, Y.; et al. Muscle cannabinoid 1 receptor regulates il-6 and myostatin expression, governing physical performance and whole-body metabolism. FASEB J. 2019, fj201801145R. [Google Scholar] [CrossRef]
- Horvath, B.; Mukhopadhyay, P.; Hasko, G.; Pacher, P. The endocannabinoid system and plant-derived cannabinoids in diabetes and diabetic complications. Am. J. Pathol. 2012, 180, 432–442. [Google Scholar] [CrossRef]
- Malenczyk, K.; Jazurek, M.; Keimpema, E.; Silvestri, C.; Janikiewicz, J.; Mackie, K.; Di Marzo, V.; Redowicz, M.J.; Harkany, T.; Dobrzyn, A. Cb1 cannabinoid receptors couple to focal adhesion kinase to control insulin release. J. Biol. Chem. 2013, 288, 32685–32699. [Google Scholar] [CrossRef]
- Kim, W.; Lao, Q.; Shin, Y.K.; Carlson, O.D.; Lee, E.K.; Gorospe, M.; Kulkarni, R.N.; Egan, J.M. Cannabinoids induce pancreatic beta-cell death by directly inhibiting insulin receptor activation. Sci. Signal. 2012, 5, ra23. [Google Scholar] [CrossRef]
- Kim, W.; Doyle, M.E.; Liu, Z.; Lao, Q.; Shin, Y.K.; Carlson, O.D.; Kim, H.S.; Thomas, S.; Napora, J.K.; Lee, E.K.; et al. Cannabinoids inhibit insulin receptor signaling in pancreatic beta-cells. Diabetes 2011, 60, 1198–1209. [Google Scholar] [CrossRef]
- Gonzalez-Mariscal, I.; Montoro, R.A.; Doyle, M.E.; Liu, Q.R.; Rouse, M.; O’Connell, J.F.; Santa-Cruz Calvo, S.; Krzysik-Walker, S.M.; Ghosh, S.; Carlson, O.D.; et al. Absence of cannabinoid 1 receptor in beta cells protects against high-fat/high-sugar diet-induced beta cell dysfunction and inflammation in murine islets. Diabetologia 2018, 61, 1470–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzechnik, A.T.; Newton, A.C. Phlpping through history: A decade in the life of phlpp phosphatases. Biochem. Soc. Trans. 2016, 44, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Kim, Y.H.; Kim, D.K.; Lee, M.W.; Lee, S.Y.; Park, T.S.; Koo, S.H.; Lee, C.H.; Choi, H.S. Activation of cannabinoid receptor type 1 (cb1r) disrupts hepatic insulin receptor signaling via cyclic amp-response element-binding protein h (crebh)-mediated induction of lipin1 gene. J. Biol. Chem. 2012, 287, 38041–38049. [Google Scholar] [CrossRef] [PubMed]
- Cinar, R.; Godlewski, G.; Liu, J.; Tam, J.; Jourdan, T.; Mukhopadhyay, B.; Harvey-White, J.; Kunos, G. Hepatic cannabinoid-1 receptors mediate diet-induced insulin resistance by increasing de novo synthesis of long-chain ceramides. Hepatology 2014, 59, 143–153. [Google Scholar] [CrossRef]
- Liu, J.; Godlewski, G.; Jourdan, T.; Liu, Z.; Cinar, R.; Xiong, K.; Kunos, G. Cannabinoid-1 receptor antagonism improves glycemic control and increases energy expenditure through sirtuin-1/mechanistic target of rapamycin complex 2 and 5’adenosine monophosphate-activated protein kinase signaling. Hepatology 2019, 69, 1535–1548. [Google Scholar] [CrossRef]
- Lee, M.W.; Chanda, D.; Yang, J.; Oh, H.; Kim, S.S.; Yoon, Y.S.; Hong, S.; Park, K.G.; Lee, I.K.; Choi, C.S.; et al. Regulation of hepatic gluconeogenesis by an er-bound transcription factor, crebh. Cell Metab. 2010, 11, 331–339. [Google Scholar] [CrossRef]
- Kim, D.K.; Kim, Y.H.; Jang, H.H.; Park, J.; Kim, J.R.; Koh, M.; Jeong, W.I.; Koo, S.H.; Park, T.S.; Yun, C.H.; et al. Estrogen-related receptor gamma controls hepatic cb1 receptor-mediated cyp2e1 expression and oxidative liver injury by alcohol. Gut 2013, 62, 1044–1054. [Google Scholar] [CrossRef]
- Kim, D.K.; Ryu, D.; Koh, M.; Lee, M.W.; Lim, D.; Kim, M.J.; Kim, Y.H.; Cho, W.J.; Lee, C.H.; Park, S.B.; et al. Orphan nuclear receptor estrogen-related receptor gamma (errgamma) is key regulator of hepatic gluconeogenesis. J. Biol. Chem. 2012, 287, 21628–21639. [Google Scholar] [CrossRef]
- Kim, D.K.; Kim, J.R.; Koh, M.; Kim, Y.D.; Lee, J.M.; Chanda, D.; Park, S.B.; Min, J.J.; Lee, C.H.; Park, T.S.; et al. Estrogen-related receptor gamma (errgamma) is a novel transcriptional regulator of phosphatidic acid phosphatase, lipin1, and inhibits hepatic insulin signaling. J. Biol. Chem. 2011, 286, 38035–38042. [Google Scholar] [CrossRef] [PubMed]
- Osei-Hyiaman, D.; DePetrillo, M.; Pacher, P.; Liu, J.; Radaeva, S.; Batkai, S.; Harvey-White, J.; Mackie, K.; Offertaler, L.; Wang, L.; et al. Endocannabinoid activation at hepatic cb1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Investig. 2005, 115, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Osei-Hyiaman, D.; Liu, J.; Zhou, L.; Godlewski, G.; Harvey-White, J.; Jeong, W.I.; Batkai, S.; Marsicano, G.; Lutz, B.; Buettner, C.; et al. Hepatic cb1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J. Clin. Investig. 2008, 118, 3160–3169. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Vemuri, V.K.; Liu, J.; Batkai, S.; Mukhopadhyay, B.; Godlewski, G.; Osei-Hyiaman, D.; Ohnuma, S.; Ambudkar, S.V.; Pickel, J.; et al. Peripheral cb1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J. Clin. Investig. 2010, 120, 2953–2966. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, T.; Demizieux, L.; Gresti, J.; Djaouti, L.; Gaba, L.; Verges, B.; Degrace, P. Antagonism of peripheral hepatic cannabinoid receptor-1 improves liver lipid metabolism in mice: Evidence from cultured explants. Hepatology 2012, 55, 790–799. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.D.; Sakurai, T.; Rainero, I.; Maj, M.C.; Kukkonen, J.P. Orexin receptor multimerization versus functional interactions: Neuropharmacological implications for opioid and cannabinoid signalling and pharmacogenetics. Pharmaceuticals 2017, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Jantti, M.H.; Putula, J.; Turunen, P.M.; Nasman, J.; Reijonen, S.; Lindqvist, C.; Kukkonen, J.P. Autocrine endocannabinoid signaling through cb1 receptors potentiates ox1 orexin receptor signaling. Mol. Pharmacol. 2013, 83, 621–632. [Google Scholar] [CrossRef]
- Zhu, F.; Wang, X.Q.; Chen, Y.N.; Yang, N.; Lang, S.Y.; Zuo, P.P.; Zhang, J.T.; Li, R.S. Changes and overlapping distribution in the expression of cb1/ox1-gpcrs in rat hippocampus by kainic acid-induced status epilepticus. Brain Res. 2015, 1597, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, K.; Thomas, M.A.; Glick, S.; Fung, E.N.; Wang, V.; Watson, L.; Gregory, P.; Antel, J.; Pelleymounter, M.A. Ibipinabant attenuates beta-cell loss in male zucker diabetic fatty rats independently of its effects on body weight. Diabetes Obes. Metab. 2012, 14, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713. [Google Scholar] [CrossRef]
- Bensaid, M.; Gary-Bobo, M.; Esclangon, A.; Maffrand, J.P.; Le Fur, G.; Oury-Donat, F.; Soubrie, P. The cannabinoid cb1 receptor antagonist sr141716 increases acrp30 mrna expression in adipose tissue of obese fa/fa rats and in cultured adipocyte cells. Mol. Pharmacol. 2003, 63, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Cinar, R.; Liu, J.; Godlewski, G.; Wesley, D.; Jourdan, T.; Szanda, G.; Mukhopadhyay, B.; Chedester, L.; Liow, J.S.; et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012, 16, 167–179. [Google Scholar] [CrossRef]
- Wang, S.; Xu, Q.; Shu, G.; Wang, L.; Gao, P.; Xi, Q.; Zhang, Y.; Jiang, Q.; Zhu, X. N-oleoyl glycine, a lipoamino acid, stimulates adipogenesis associated with activation of cb1 receptor and akt signaling pathway in 3t3-l1 adipocyte. Biochem. Biophys. Res. Commun. 2015, 466, 438–443. [Google Scholar] [CrossRef]
- Nagappan, A.; Jung, D.Y.; Kim, J.H.; Jung, M.H. Protective effects of gomisin n against hepatic cannabinoid type 1 receptor-induced insulin resistance and gluconeogenesis. Int. J. Mol. Sci. 2018, 19, 968. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl.No. | Localization | Biological Role | Implication |
|---|---|---|---|
| 1 | Brain | Anxiety and depression, appetite and food intake, reward and addiction, neuroprotection, neural development, and sleep | CB1R overexpression results in increased food intake and induces overeating. |
| 2 | Liver | Liver steatosis, fibrosis, insulin resistance, lipogenesis, splanchnic, and vasodilation | CB1R overexpression induces insulin resistance and obesity. |
| 3 | Cardiovascular system | Cardiac function, negative inotropy, and vasodilation | CB1R overexpression promotes cardiomyocyte injury, oxidative stress, inflammation, and fibrosis. |
| 4 | Skeletal muscle | Energy metabolism and muscle fiber formation | CB1R overexpression impairs insulin signaling. |
| 5 | Reproductive system | Fertility regulation, embryo implantation, and embryonic development | Activation of CB1R modulates the oocyte maturation and embryonic development. |
| 6 | GI tract | GI motility, enteroendocrine function, and energy balance. | CB1R agonist reduces GI motility and colitis-related inflammation. |
| Sl.No. | Compounds | Biological Benefits | Cell Models |
|---|---|---|---|
| 1 | SR141716 | Antiobesity, hepatoprotective, promoted insulin sensitivity, reduced aging-related insulin resistance, and reduced metabolic dysfunction | Mouse 3T3 F442A cells, obese (fa/fa) rats, 3T3-L1 adipocytes, rat L6 myotubes, and C57BL/6 mice |
| 2 | AM6545 | Cardiometabolic risk | HFD-fed obese mice and CB1R−/− transgenic mice |
| 3 | JD5037 | Antiobesity effects by reversing leptin resistance | Diet-induced obesity (DIO) mice |
| 4 | Ibipinabant | Antidiabetic effects | β-cells |
| 5 | Gomisin N | Reduced insulin resistance and gluconeogenesis | HepG2 cells |
| 6 | N-oleoyl glycine | Reduced adipogenesis and increased insulin sensitivity | 3T3-L1 adipocytes |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagappan, A.; Shin, J.; Jung, M.H. Role of Cannabinoid Receptor Type 1 in Insulin Resistance and Its Biological Implications. Int. J. Mol. Sci. 2019, 20, 2109. https://doi.org/10.3390/ijms20092109
Nagappan A, Shin J, Jung MH. Role of Cannabinoid Receptor Type 1 in Insulin Resistance and Its Biological Implications. International Journal of Molecular Sciences. 2019; 20(9):2109. https://doi.org/10.3390/ijms20092109
Chicago/Turabian StyleNagappan, Arulkumar, Jooyeon Shin, and Myeong Ho Jung. 2019. "Role of Cannabinoid Receptor Type 1 in Insulin Resistance and Its Biological Implications" International Journal of Molecular Sciences 20, no. 9: 2109. https://doi.org/10.3390/ijms20092109
APA StyleNagappan, A., Shin, J., & Jung, M. H. (2019). Role of Cannabinoid Receptor Type 1 in Insulin Resistance and Its Biological Implications. International Journal of Molecular Sciences, 20(9), 2109. https://doi.org/10.3390/ijms20092109