Characterization and Rapid Gene-Mapping of Leaf Lesion Mimic Phenotype of spl-1 Mutant in Soybean (Glycine max (L.) Merr.)

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Characterization of spl-1 Mutant
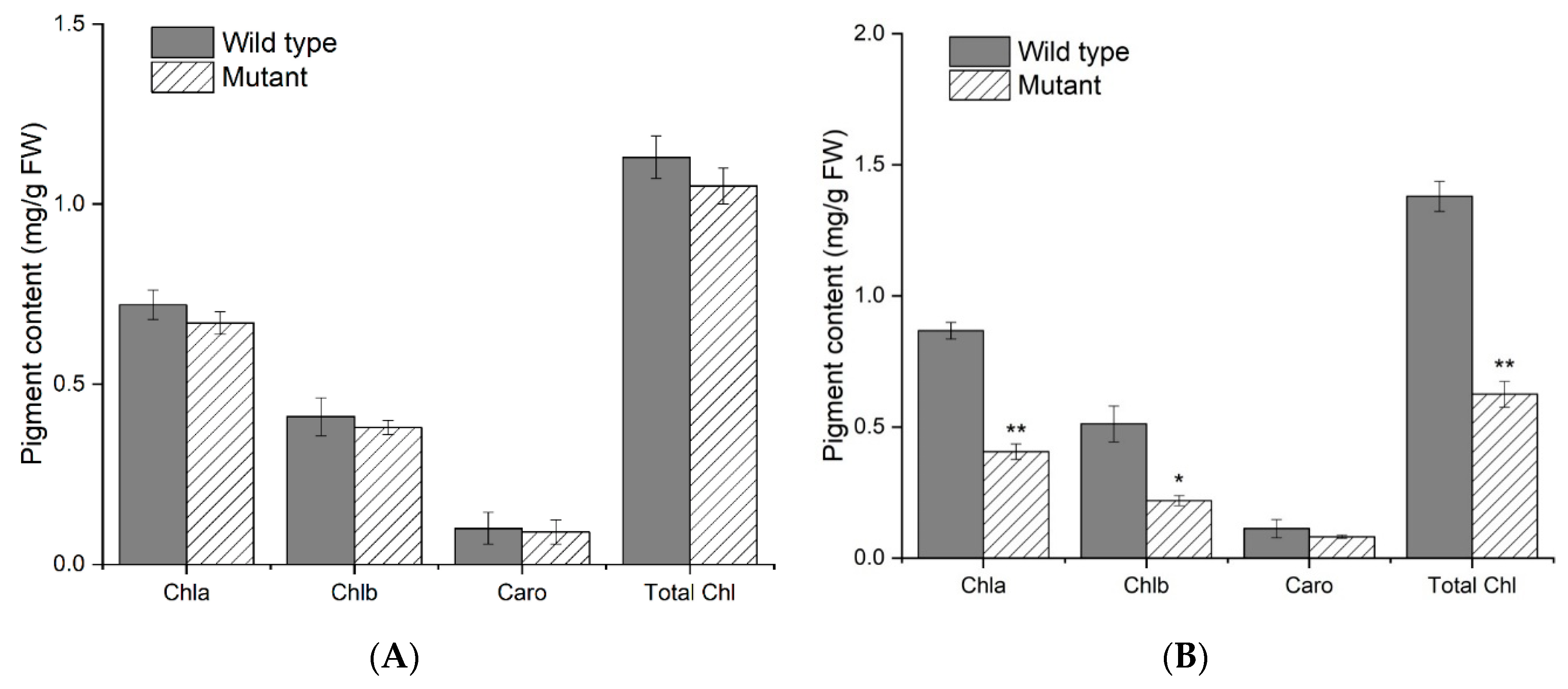
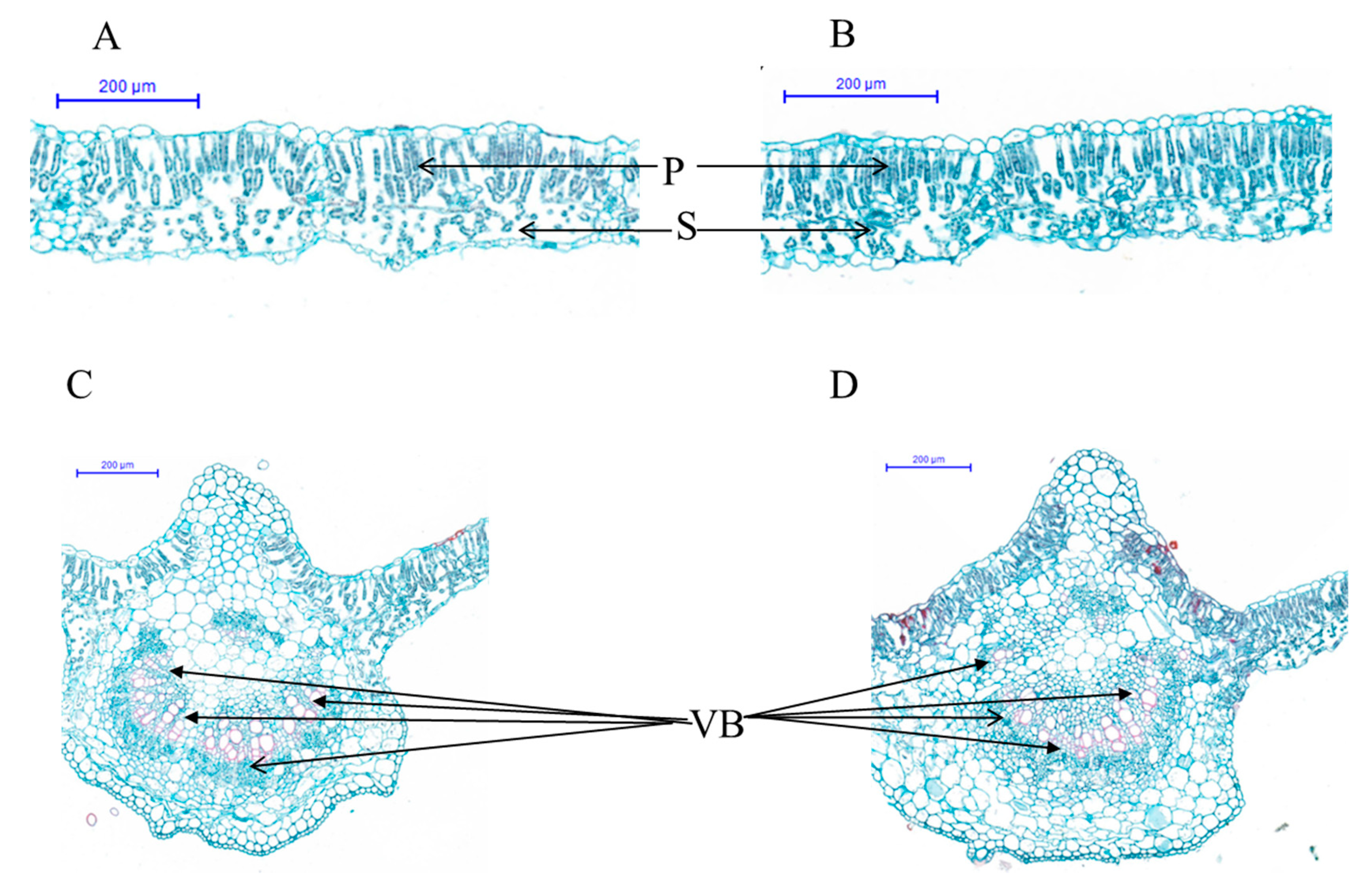
2.2. Leaf Pigment Content and Histological Analysis
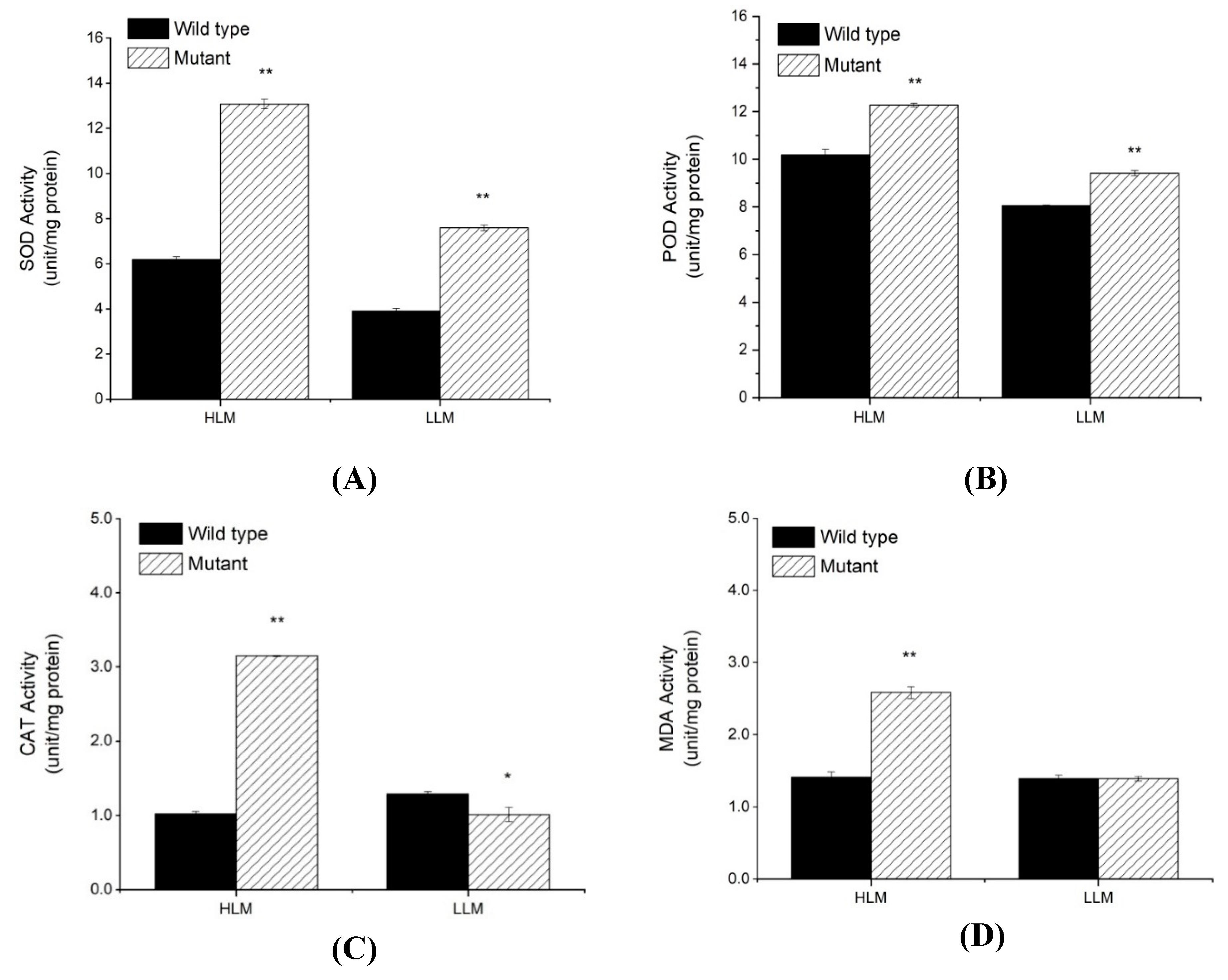
2.3. Physiochemical Analysis for PCD, H2O2 Accumulation and Antioxidants
2.4. Inheritance for Spotted Leaf Trait of spl-1 Mutant
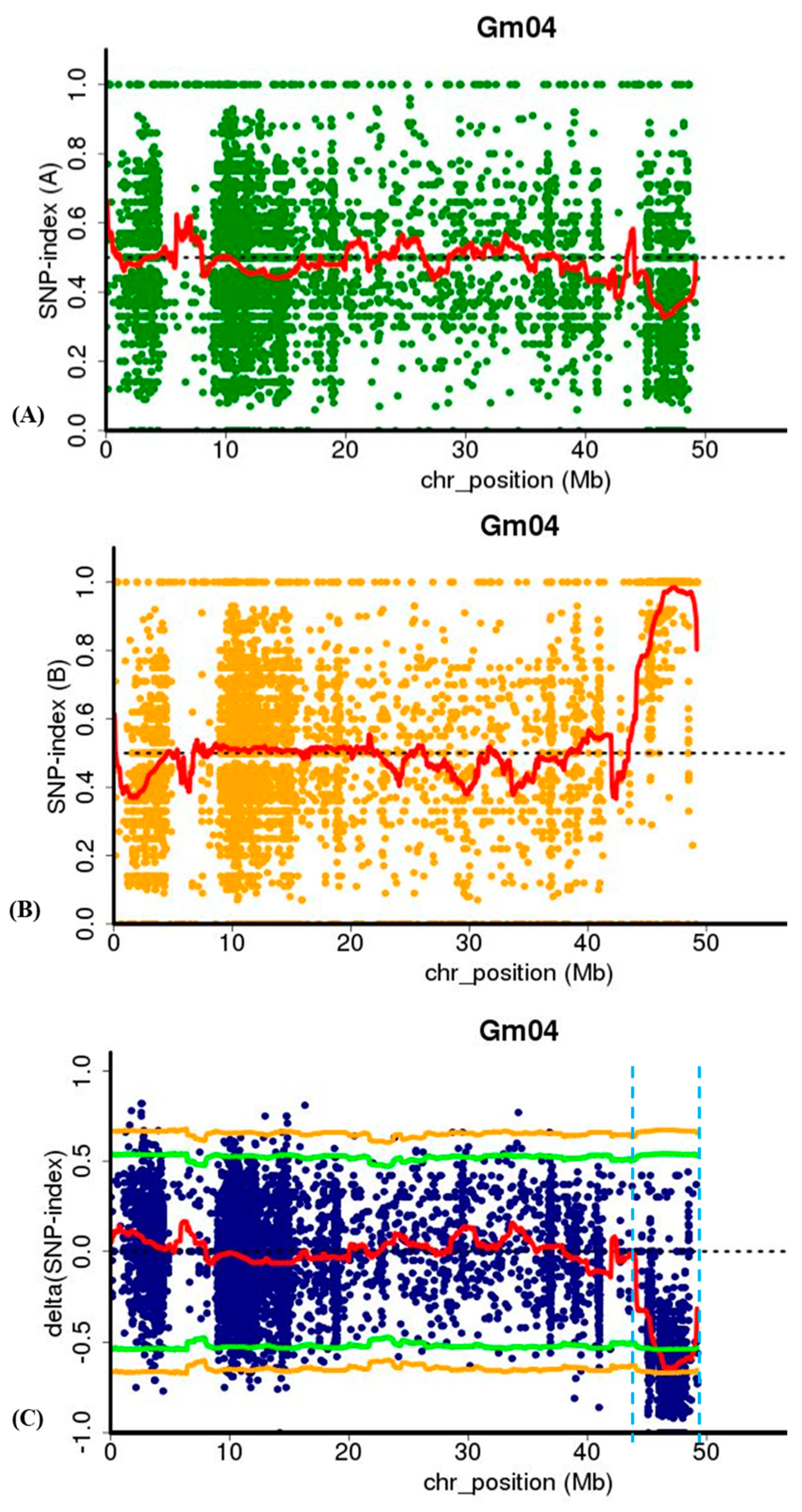
2.5. MutMap Analysis for Identification of lm1 Locus
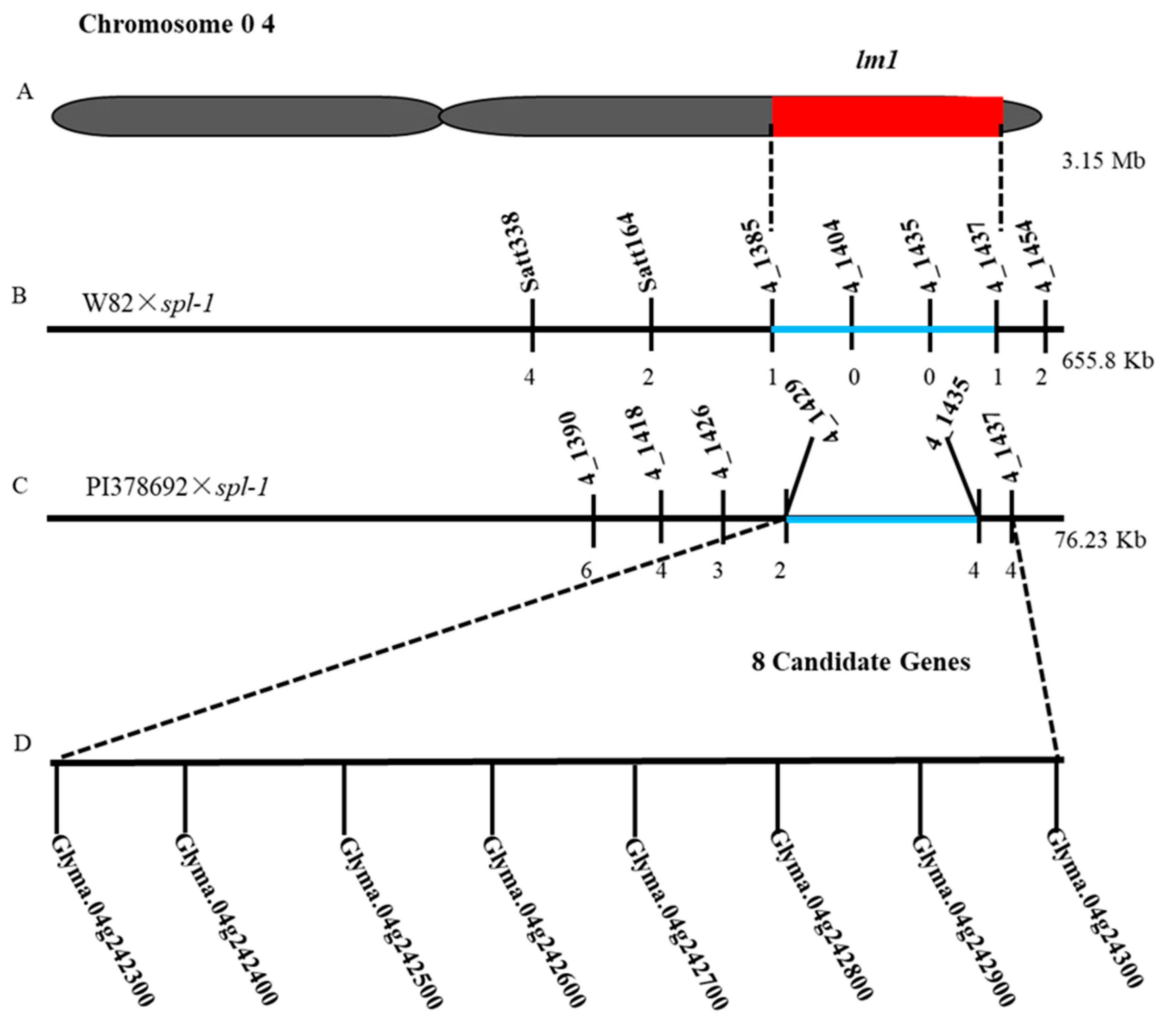
2.6. Validation and Fine Mapping of the lm1 Locus
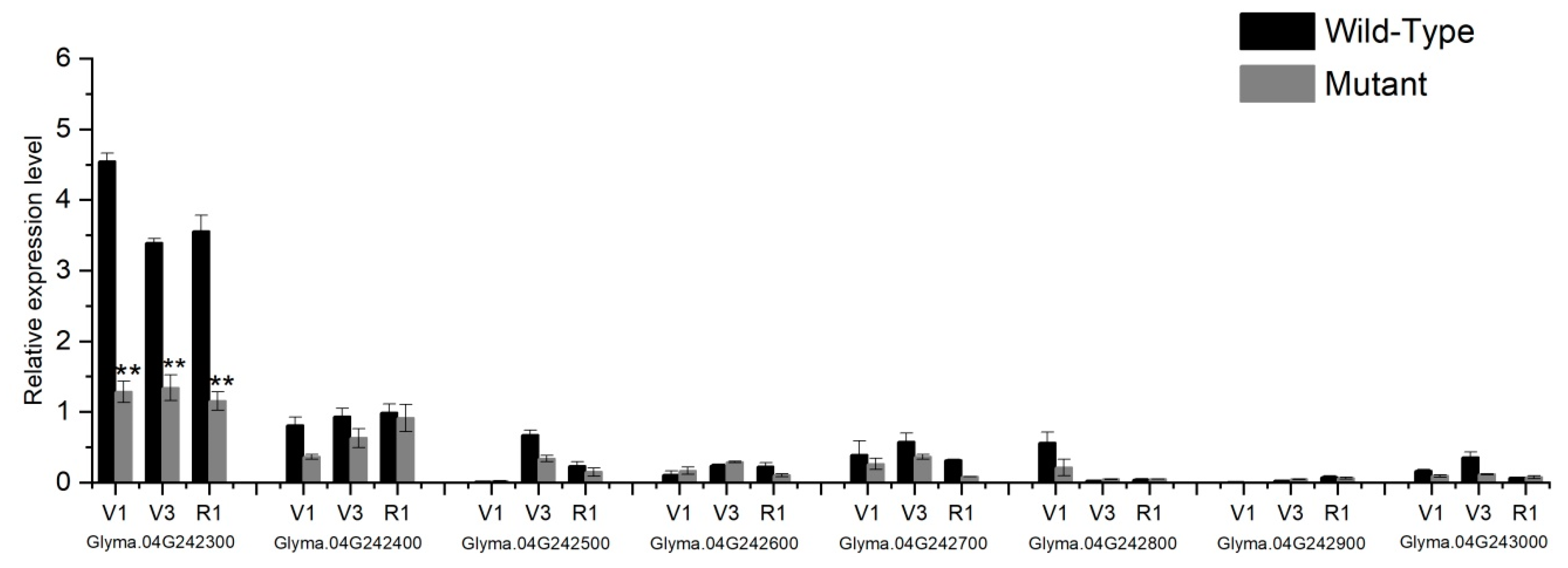
2.7. qRT-PCR Expression and Sequences Analysis of Candidate Genes
3. Discussion
3.1. spl-1 Is a New Soybean Leaf Lesion Mutant with Special Characteristics
3.2. Deploying MutMap and Traditional Mapping Methods to Accelerate Gene Identification
3.3. Candidate Genes for spl-1 Phenotype
4. Materials and Methods
4.1. Plant Materials and Phenotypic Evaluation
4.2. Leaf Pigment Quantification and Histological Analyses
4.3. Leaf Histochemical and Physiological Analyses
4.3.1. Trypan Blue Staining of Cell Death
4.3.2. H2O2 Detection by DAB
4.3.3. Antioxidants Activities and Lipid Peroxidation Determination
4.4. MutMap Analysis
4.4.1. Construction of MutMap Libraries and Illumina Sequencing
4.4.2. SNP Index Analysis
4.4.3. Sliding-Window Analysis
4.5. Fine Mapping of lm1 Locus
4.6. Expression and Sequence Analysis of Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LMMs | lesion mimic mutants |
| spl-1 | spotted Leaf-1 |
| lm1 | lesion mimic 1 |
| PCD | programmed cell death |
| HR | hypersensitive response |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| POD | peroxidase |
| CAT | catalase |
| MDA | malondialdehyde |
| SSR | simple sequence repeat |
| PLC | plantacyanin |
| PC | phytocyanin |
| qRT-PCR | quantitative real-time PCR |
| NGS | next-generation sequencing |
| WGRS | whole genome re-sequencing |
| DAB | 3,3′-diaminobenzidine |
| SNP | Single Nucleotide Polymorphisms |
References
- Li, R.; Chen, S.; Liu, G.; Han, R.; Jiang, J. Characterization and identification of a woody lesion mimic mutant lmd, showing defence response and resistance to Alternaria alternate in birch. Sci. Rep. 2017, 7, 11308. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, Z.; Liu, T.; Gao, B.; Xiong, X. Identification and map-based cloning of the Light-Induced Lesion Mimic Mutant 1 (LIL1) gene in rice. Front. Plant Sci. 2017, 8, 2122. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hu, H.; Zhang, Y.; Wang, Z.; Dong, G.; Cui, Y.; Qian, Q.; Ren, D.; Guo, L. Genetic analysis and fine-mapping of a new rice mutant, white and lesion mimic leaf1. Plant Growth Regul. 2018, 85, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.P.; Wen, R.; Wang, J.H.; Xiang, D.Q.; Wang, Q.; Zang, Y.P.; Wang, Z.; Huang, S.; Li, X.; Datla, R.; et al. Arabidopsis UBC13 differentially regulates two programmed cell death pathways in responses to pathogen and low-temperature stress. New Phytol. 2019, 221, 919–934. [Google Scholar] [CrossRef]
- Dietrich, R.A.; Delaney, T.P.; Uknes, S.J.; Ward, E.R.; Ryals, J.A.; Dangl, J.L. Arabidopsis mutants simulating disease resistance response. Cell 1994, 77, 565–577. [Google Scholar] [CrossRef]
- Badigannavar, A.M.; Kale, D.M.; Eapen, S.; Murty, G.S.S. Inheritance of disease lesion mimic leaf trait in groundnut. J. Hered. 2002, 93, 50–52. [Google Scholar] [CrossRef]
- Walbot, V. Maize Mutants for the 21st Century. Plant Cell 1991, 3, 851–856. [Google Scholar] [CrossRef]
- Lorrain, S.; Vailleau, F.; Balaque, C.; Roby, D. Lesion mimic mutants: Keys for deciphering cell death and defense pathways in plants? Trends Plant Sci. 2003, 8, 263–271. [Google Scholar] [CrossRef]
- Mizobuchi, R.; Hirabayashi, H.; Kaji, R.; Nishizawa, Y.; Satoh, H.; Ogawa, T.; Okamoto, M. Differential expression of disease resistance in rice lesion-mimic mutants. Plant Cell Rep. 2002, 2, 390–396. [Google Scholar] [CrossRef]
- Wu, C.J.; Bordeos, A.; Madamba, M.R.S.; Baraoidan, M.; Ramos, M.; Wang, G.L.; Leach, J.E.; Leung, H. Rice lesion mimic mutants with enhanced resistance to diseases. Mol. Genet. Genom. 2008, 279, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Buschges, R.; Hollricher, K.; Panstruga, R.; Simons, G.; Wolter, M.; Frijters, A.; van Daelen, R.; van der Lee, T.; Diergaarde, P.; Groenendijk, J.; et al. The barley mlo gene: A novel control element of plant pathogen resistance. Cell 1997, 88, 695–705. [Google Scholar] [CrossRef]
- Rostoks, N.; Schmierer, D.; Mudie, S.; Drader, T.; Brueggeman, R.; Caldwell, D.G.; Waugh, R.; Kleinhofs, A. Barley necrotic locus nec1 encodes the cyclic nucleotide-gated ion channel 4 homologous to the Arabidopsis HLM1. Mol. Genet. Genom. 2006, 275, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Johal, G.S.; Hulbert, S.H.; Briggs, S.P. Disease lesion mimics of maize: A model for cell death in plants. Bioessays 1995, 17, 685–692. [Google Scholar] [CrossRef]
- Chung, J.; Staswick, P.E.; Graef, G.L.; Wysong, D.S.; Specht, J.E. Inheritance of a disease lesion mimic mutant in soybean. J. Hered. 1998, 89, 363–365. [Google Scholar] [CrossRef]
- Kim, H.K.; Kim, J.K.; Paek, K.B.; Kim, Y.J.; Chung, J. The phenotype of the soybean disease-lesion imic (dlm) mutant is light-dependentand associated with chloroplast function. Plant Path. J. 2005, 21, 395–401. [Google Scholar] [CrossRef]
- Moeder, W.; Yoshioka, K. Lesion mimic mutants A classical, yet still fundamental approach to study programmed cell death. Plant Sig. Behav. 2008, 3, 764–767. [Google Scholar] [CrossRef]
- Jeong, W.H.; Lee, K.J.; Park, M.S.; Nam, K.C.; Kim, M.S.; Chung, J.I. Independent Inheritance of dlm Allele with lf2 and P1 alleles in soybean (Glycine max L.). Korean J. Breed. Sci. 2007, 39, 232–235. [Google Scholar]
- Sung, M.K.; Kim, M.H.; Seo, H.J.; Chung, J.I. Inheritance of dlm and ti genes in soybean. Plant Breed. Biotechnol. 2013, 1, 9–13. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, W.; Zhang, Y.; Liu, M.; Kong, J.; Yu, Z.; Jaffer, A.M.; Gai, J.; Zhao, T. Identification of two duplicated loci controlling a disease-like rugose leaf phenotype in soybean. Crop Sci. 2016, 56, 1611–1618. [Google Scholar] [CrossRef]
- Lu, P.; Qin, J.; Wang, G.; Wang, L.; Wang, Z.; Wu, Q.; Xie, J.; Liang, Y.; Wang, Y.; Zhang, D.; et al. Comparative fine mapping of the Wax 1 (W1) locus in hexaploid wheat. Theor. Appl. Genet. 2015, 128, 1595–1603. [Google Scholar] [CrossRef]
- Xu, J.; Wang, B.; Wu, Y.; Du, P.; Wang, J.; Wang, M.; Yi, C.; Gu, M.; Liang, G. Fine mapping and candidate gene analysis of ptgms2-1, the photoperiod-thermo-sensitive genic male sterile gene in rice (Oryza sativa L.). Theor. Appl. Genet. 2011, 122, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Hu, J.; Xu, J.; Yu, H.; Shi, Z.; Xiong, G.; Zhu, L.; Zeng, D.; Zhang, G.; Gao, Z.; et al. Identification and characterization of Mini1, a gene regulating rice shoot development. J. Integr. Plant Biol. 2015, 57, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Li, Z.; Liu, Z.; Guo, Y.; Qiu, L. Next-generation sequencing from Bulked-Segregant Analysis accelerates the simultaneous identification of two qualitative genes in soybean. Front. Plant Sci. 2017, 8, 919. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, K.; Weigel, D. Fast-forward genetics enabled by new sequencing technologies. Trends Plant Sci. 2011, 16, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Zegeye, W.A.; Zhang, Y.; Cao, L.; Cheng, S. Whole Genome Resequencing from bulked populations as a rapid QTL and gene identification method in rice. Inter. J. Mol. Sci. 2018, 19, 4000. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Upadhyaya, H.D.; Bajaj, D.; Kujur, A.; Badoni, S.; Laxmi; Kumar, V.; Tripathi, S.; Gowda, C.L.L.; Sharma, S.; et al. Deploying QTL-seq for rapid delineation of a potential candidate gene underlying major trait-associated QTL in chickpea. DNA Res. 2015, 22, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascher, M.; Jost, M.; Kuon, J.E.; Himmelbach, A.; Aßfalg, A.; Beier, S.; Scholz, U.; Graner, A.; Stein, N. Mapping-by-sequencing accelerates forward genetics in barley. Genome Biol. 2014, 15, R78. [Google Scholar] [CrossRef]
- Liu, S.; Yeh, C.T.; Tang, H.M.; Nettleton, D.; Schnable, P.S. Gene mapping via Bulked Segregant RNA-Seq (BSR-Seq). PLoS ONE 2012, 7, e36406. [Google Scholar] [CrossRef]
- Zhong, C.; Sun, S.; Li, Y.; Duan, C.; Zhu, Z. Next-generation sequencing to identify candidate genes and develop diagnostic markers for a novel Phytophthora resistance gene, RpsHC18, in soybean. Theor. Appl. Genet. 2018, 131, 525–538. [Google Scholar] [CrossRef]
- Illa-Berenguer, E.; Houten, J.V.; Huang, Z.; van der Knaap, E. Rapid and reliable identification of tomato fruit weight and locule number loci by QTL-seq. Theor. Appl. Genet. 2015, 128, 1329–1342. [Google Scholar] [CrossRef]
- Lu, H.; Lin, T.; Klein, J.; Wang, S.; Qi, J.; Zhou, Q.; Sun, J.; Zhang, Z.; Weng, Y.; Huang, S. QTL-seq identifies an early flowering QTL located near flowering locus T in cucumber. Theor. Appl. Genet. 2014, 127, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Pruyn, M.L.; Spicer, R. Parenchyma. e LS 2001. [Google Scholar] [CrossRef]
- Kura-Hotta, M.; Satoh, K.; Katoh, S. Relationship between photosynthesis and chlorophyll content during leaf senescence of rice seedlings. Plant Cell Physiol. 1987, 28, 1321–1329. [Google Scholar]
- Sack, L.; Cowan, P.D.; Jaikumar, N.; Holbrook, N.M. The ‘hydrology’ of leaves: Co-ordination of structure and function in temperate woody species. Plant Cell Environ. 2003, 26, 1343–1356. [Google Scholar] [CrossRef]
- Sack, L.; Holbrook, N.M. Leaf hydraulics. Annu. Rev. Plant Biol. 2006, 57, 361–381. [Google Scholar] [CrossRef]
- Brodribb, T.J.; Feild, T.S.; Sack, L. Viewing leaf structure and evolution from a hydraulic perspective. Funct. Plant Biol. 2010, 37, 488–498. [Google Scholar] [CrossRef]
- Brodribb, T.J.; Holbrook, N.M. Stomatal closure during leaf dehydration, correlation with other leaf physiological traits. Plant Physiol. 2003, 132, 2166–2173. [Google Scholar] [CrossRef]
- Wang, J.; Ye, B.; Yin, J.; Yuan, C.; Zhou, X.; Li, W.; He, M.; Wang, J.; Chen, W.; Qin, P.; et al. Characterization and fine mapping of a light-dependent leaf lesion mimic mutant 1 in rice. Plant Physiol. Biochem. 2015, 97, 44–51. [Google Scholar] [CrossRef]
- Van Breusegem, F.; Dat, J.F. Reactive Oxygen Species in Plant Cell Death. Plant Physiol. 2006, 141, 384–390. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Dong, J.; Kim, S.T.; Lord, E.M. Plantacyanin plays a role in reproduction in Arabidopsis. Plant Physiol. 2005, 138, 778–789. [Google Scholar] [CrossRef]
- Fehr, W.R.; Caviness, C.E.; Burmood, D.T.; Pennington, J.S. Stage of development descriptions for soybeans, Glycine max (L.) Merrill. Crop Sci. 1971, 11, 929–931. [Google Scholar] [CrossRef]
- Hunt, M.D.; Neuenschwander, U.H.; Delaney, T.P.; Weymann, K.B.; Friedrich, L.B.; Lawton, K.A.; Steiner, H.Y.; Ryals, J.A. Recent advances in systemic acquired resistance research—A review. Gene 1996, 179, 89–95. [Google Scholar] [CrossRef]
- Yin, Z.C.; Chen, J.; Zeng, L.R.; Goh, M.L.; Leung, H.; Khush, G.S.; Wang, G.L. Characterizing rice lesion mimic mutants and identifying a mutant with broad-spectrum resistance to rice blast and bacterial blight. Mol. Plant-Microbe Interact. 2000, 13, 869. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, N.; Tanaka, R.; Grimm, B.; Masuda, T.; Moulin, M.; Smith, A.G.; Tanaka, A.; Terry, M.J. The cell biology of tetrapyrroles: A life and death struggle. Trends Plant Sci. 2010, 15, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Quesada, V.; Sarmiento-Manus, R.; Gonzalez-Bayon, R.; Hricova, A.; Ponce, M.R.; Micol, J.L. PORPHOBILINOGEN DEAMINASE deficiency alters vegetative and reproductive development and causes lesions in arabidopsis. PLoS ONE 2013, 8, e53378. [Google Scholar] [CrossRef] [PubMed]
- Meskauskiene, R.; Nater, M.; Goslings, D.; Kessler, F.; den Camp, R.O.; Apel, K. FLU: A negative regulator of chlorophyll biosynthesis in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2001, 98, 12826–12831. [Google Scholar] [CrossRef]
- Samol, I.; Buhr, F.; Springer, A.; Pollmann, S.; Lahroussi, A.; Rossig, C.; von Wettstein, D.; Reinbothe, C.; Reinbothe, S. Implication of the oep16-1 Mutation in a flu-Independent, singlet oxygen-regulated cell death pathway in Arabidopsis thaliana. Plant Cell Physiol. 2011, 52, 84–95. [Google Scholar] [CrossRef]
- Hu, G.S.; Yalpani, N.; Briggs, S.P.; Johal, G.S. A porphyrin pathway impairment is responsible for the phenotype of a dominant disease lesion mimic mutant of maize. Plant Cell 1998, 10, 1095–1105. [Google Scholar] [CrossRef]
- Ishikawa, A.; Okamoto, H.; Iwasaki, Y.; Asahi, T. A deficiency of coproporphyrinogen III oxidase causes lesion formation in Arabidopsis. Plant J. 2001, 27, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Liu, L.; Tang, J.; Lin, A.; Zhang, F.; Fang, J.; Zhang, G.; Chu, C. RLIN1, encoding a putative coproporphyrinogen III oxidase, is involved in lesion initiation in rice. J. Genet. Genom. 2011, 38, 29–37. [Google Scholar] [CrossRef]
- Greenberg, J.T.; Ausubel, F.M. Arabidopsis mutants compromised for the control of cellular damage during pathogenesis and aging. Plant J. 1993, 4, 327–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mach, J.M.; Castillo, A.R.; Hoogstraten, R.; Greenberg, J.T. The Arabidopsis accelerated cell death gene ACD2 encodes red chlorophyll catabolite reductase and suppresses the spread of disease symptoms. Proc. Natl. Acad. Sci. USA 2001, 98, 771–776. [Google Scholar] [CrossRef]
- Tanaka, R.; Hirashima, M.; Satoh, S.; Tanaka, A. The Arabidopsis-accelerated cell death gene ACD1 is involved in oxygenation of pheophorbide a: Inhibition of the pheophorbide a oxygenase activity does not lead to the “stay-green” phenotype in Arabidopsis. Plant Cell Physiol. 2003, 44, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Schmelz, E.A.; Muthukrishnan, S. Development of a lesion-mimic phenotype in a transgenic wheat line overexpressing genes for pathogenesis-related (PR) proteins is dependent on salicylic acid concentration. Mol. Plant-Microbe Interact. 2003, 16, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Lin, Q.; Zhu, L.; Ren, Y.; Zhou, K.; Shabek, N.; Wu, F.; Mao, H.; Dong, W.; Gan, L.; et al. D14-SCFD3-dependent degradation of D53 regulates strigolactone signalling. Nature 2013, 504, 406. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Yu, T.; Gao, S.; Xiong, C.; Xie, Q.; Li, H.; Ye, Z.; Yang, C. Fine mapping of the dialytic gene that controls multicellular trichome formation and stamen development in tomato. Theor. Appl. Genet. 2016, 129, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.J.; Watanabe, S.; Yamada, T.; Tsubokura, Y.; Nakashima, H.; Zhai, H.; Anai, T.; Sato, S.; Yamazaki, T.; Lu, S.X.; et al. Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc. Natl. Acad. Sci. USA 2012, 109, E2155–E2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wang, W.; Guo, N.; Zhang, Y.; Bu, Y.; Zhao, J.; Xing, H. Combining QTL-seq and linkage mapping to fine map a wild soybean allele characteristic of greater plant height. BMC Genom. 2018, 19, 226. [Google Scholar] [CrossRef] [PubMed]
- Kadambari, G.; Vemireddy, L.R.; Srividhya, A.; Nagireddy, R.; Jena, S.S.; Gandikota, M.; Patil, S.; Veeraghattapu, R.; Deborah, D.A.K.; Reddy, G.E.; et al. QTL-Seq-based genetic analysis identifies a major genomic region governing dwarfness in rice (Oryza sativa L.). Plant Cell Rep. 2018, 37, 677–689. [Google Scholar] [CrossRef]
- Ryden, L.G.; Hunt, L.T. Evolution of protein complexity - the blue copper-containing oxidases and related proteins. J. Mol. Evol. 1993, 36, 41–66. [Google Scholar] [CrossRef]
- Roach, T.; Krieger-Liszkay, A. Regulation of photosynthetic electron transport and photoinhibition. Curr. Protein Pept. Sci. 2014, 15, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.P.; Yu, Y.; Feng, Y.Z.; Zhou, Y.F.; Zhang, F.; Yang, Y.W.; Lei, M.Q.; Zhang, Y.C.; Chen, Y. MiR408 pregulates grain yield and photosynthesis via a phytocyanin rotein. Plant Physiol. 2017, 175, 1175–1185. [Google Scholar] [CrossRef]
- Fedorova, M.; van de Mortel, J.; Matsumoto, P.A.; Cho, J.; Town, C.D.; VandenBosch, K.A.; Gantt, J.S.; Vance, C.P. Genome-wide identification of nodule-specific transcripts in the model legume Medicago truncatula. Plant Physiol. 2002, 130, 519–537. [Google Scholar] [CrossRef]
- Kim, S.; Mollet, J.C.; Dong, J.; Zhang, K.L.; Park, S.Y.; Lord, E.M. Chemocyanin, a small basic protein from the lily stigma, induces pollen tube chemotropism. Proc. Natl. Acad. Sci. USA 2003, 100, 16125–16130. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Zhao, H.; Liu, Z.; Zhao, J. The Phytocyanin Gene Family in Rice (Oryza sativa L.): Genome-wide identification, classification and transcriptional analysis. PLoS ONE 2011, 6, e25184. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, Z.N.; Talame, V.; Deyholos, M.; Michalowski, C.B.; Galbraith, D.W.; Gozukirmizi, N.; Tuberosa, R.; Bohnert, H.J. Monitoring large-scale changes in transcript abundance in drought- and salt-stressed barley. Plant Mol. Biol. 2002, 48, 551–573. [Google Scholar] [CrossRef]
- Kreps, J.A.; Wu, Y.J.; Chang, H.S.; Zhu, T.; Wang, X.; Harper, J.F. Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 2002, 130, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Provart, N.J.; Gil, P.; Chen, W.Q.; Han, B.; Chang, H.S.; Wang, X.; Zhu, T. Gene expression phenotypes of Arabidopsis associated with sensitivity to low temperatures. Plant Physiol. 2003, 132, 893–906. [Google Scholar] [CrossRef]
- Chen, X.M. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 2004, 303, 2022–2025. [Google Scholar] [CrossRef]
- Llave, C.; Xie, Z.X.; Kasschau, K.D.; Carrington, J.C. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 2002, 297, 2053–2056. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Zhu, J.K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, H.K. Chlorophylls and carotenoids-pigments of photosynthetic biomembranes. Methods Enzymol. 1987, 148, 350–382. [Google Scholar]
- Carland, F.M.; McHale, N.A. LOP1: A gene involved in auxin transport and vascular patterning in Arabidopsis. Development 1996, 122, 1811–1819. [Google Scholar]
- Chen, F.; Wang, F.; Wu, F.; Mao, W.; Zhang, G.; Zhou, M. Modulation of exogenous glutathione in antioxidant defense system against Cd stress in the two barley genotypes differing in Cd tolerance. Plant Physiol. Biochem. 2010, 48, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Nahar, K.; Hasanuzzaman, M.; Fujita, M. Calcium supplementation improves Na+/K+ ratio, antioxidant defense and glyoxalase systems in salt-stressed rice seedlings. Front. Plant Sci. 2016, 7, 609. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Liu, L.; Liu, Q.; Bi, Z.; Yu, N.; Cheng, S.; Cao, L. Fine mapping of the lesion mimic and early senescence 1 (lmes1) in rice (Oryza sativa). Plant Physiol. Biochem. 2014, 80, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Takagi, H.; Uemura, A.; Yaegashi, H.; Tamiru, M.; Abe, A.; Mitsuoka, C.; Utsushi, H.; Natsume, S.; Kanzaki, H.; Matsumura, H.; et al. MutMap-Gap: Whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol. 2013, 200, 276–283. [Google Scholar] [CrossRef]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Fekih, R.; Takagi, H.; Tamiru, M.; Abe, A.; Natsume, S.; Yaegashi, H.; Sharma, S.; Sharma, S.; Kanzaki, H.; Matsumura, H.; et al. MutMap plus: Genetic mapping and mutant identification without crossing in rice. PLoS ONE 2013, 8, e68529. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Khan, A.W.; Saxena, R.K.; Kumar, V.; Kale, S.M.; Sinha, P.; Chitikineni, A.; Pazhamala, L.T.; Garg, V.; Sharma, M.; et al. Next-generation sequencing for identification of candidate genes for Fusarium wilt and sterility mosaic disease in pigeonpea (Cajanus cajan). Plant Biotechnol. J. 2016, 14, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.J.; Jia, G.F.; Zhu, Y.L.; Grant, D.; Nelson, R.T.; Hwang, E.Y.; Hyten, D.L.; Cregan, P.B. Abundance of SSR motifs and development of candidate polymorphic SSR markers (BARCSOYSSR_1.0) in soybean. Crop Sci. 2010, 50, 1950–1960. [Google Scholar] [CrossRef]
- Michelmore, R.W.; Paran, I.; Kesseli, R.V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef]
- Lander, E.S.; Green, P.; Abrahamson, J.; Barlow, A.; Daly, M.J.; Lincoln, S.E.; Newberg, L.A. MAPMAKER: An interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1987, 1, 1174–1181. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cross. | Generation | No. of Plants/Lines | Expected Ratio | χ2 | p | |||
|---|---|---|---|---|---|---|---|---|
| Total | Wild Type | Segregation | Mutant | |||||
| W82×spl-1 | F2 | 400 | 310 | - | 90 | 3:1 | 1.20 | 0.27 |
| F2:3 line | 20 | 6 | 14 | 0 | 1:2 | 0.01 | 0.94 | |
| PI378692×spl-1 | F2 | 793 | 609 | - | 184 | 3:1 | 1.27 | 0.26 |
| F2:3 line | 13 | 4 | 9 | 0 | 1:2 | 0.01 | 0.92 | |
| Marker | Chromosome | Start * | End | Primer (F/R) Sequences | |
|---|---|---|---|---|---|
| BARCSOYSSR_04_1390 | Gm04 | 50549768 | 50549787 | F | CCCGGTACAGTTGAGATGGA |
| 50550014 | 50549995 | R | TTGCACTTCAGTAGGCCCTC | ||
| BARCSOYSSR_04_1391 | Gm04 | 50588500 | 50588519 | F | AGATGGTGGTGTTCTCAGGG |
| 50588766 | 50588747 | R | ACCATCACCAACATGCAGAT | ||
| BARCSOYSSR_04_1418 | Gm04 | 50942037 | 50942061 | F | TTTTTCTTCAGAAACTTGAAACATT |
| 50942254 | 50942234 | R | TGCATTTCTGAAACAAGGCAT | ||
| BARCSOYSSR_04_1420 | Gm04 | 50953149 | 50953174 | F | AAGTGATCAATGTTATCGATGAAGTA |
| 50953433 | 50953409 | R | TTTGTCTCAATTAGTGTGGAATTTG | ||
| BARCSOYSSR_04_1426 | Gm04 | 51011052 | 51011071 | F | ATCAGAGGTCTGCCACCAAT |
| 51011271 | 51011252 | R | CGCTGACAGACACCAAGAGA | ||
| BARCSOYSSR_04_1429 | Gm04 | 51035485 | 51035504 | F | TTTGCTACAGTGCTATCGGC |
| 51035766 | 51035747 | R | TGCCAGCCGCTTATCTATCT | ||
| BARCSOYSSR_04_1435 | Gm04 | 51111716 | 51111735 | F | GTCCGTGCCAGTTTTTCATT |
| 51111960 | 51111941 | R | TGCTGCACTTTCTCCTGATG | ||
| Gene ID | Position (bp) | Direction | Function Annotation |
|---|---|---|---|
| Glyma.04g242300 | 51036742-51037897 | Forward | Plantacyanin |
| Glyma.04g242400 | 51047485-51048880 | Forward | Unknown |
| Glyma.04g242500 | 51053346-51056055 | Reverse | Flavin-binding monooxygenase family protein |
| Glyma.04g242600 | 51061920-51064096 | Forward | Unknown |
| Glyma.04g242700 | 51064744-51067380 | Reverse | F-box/RNI-like superfamily protein |
| Glyma.04g242800 | 51082744-51092913 | Reverse | ACT domain repeat 3 |
| Glyma.04g242900 | 51103163-51108385 | Reverse | Protein kinase superfamily protein |
| Glyma.04g243000 | 51109534-51115501 | Reverse | Thiamin diphosphate-binding fold (THDP-binding) superfamily protein |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Amin, G.M.; Kong, K.; Sharmin, R.A.; Kong, J.; Bhat, J.A.; Zhao, T. Characterization and Rapid Gene-Mapping of Leaf Lesion Mimic Phenotype of spl-1 Mutant in Soybean (Glycine max (L.) Merr.). Int. J. Mol. Sci. 2019, 20, 2193. https://doi.org/10.3390/ijms20092193
Al Amin GM, Kong K, Sharmin RA, Kong J, Bhat JA, Zhao T. Characterization and Rapid Gene-Mapping of Leaf Lesion Mimic Phenotype of spl-1 Mutant in Soybean (Glycine max (L.) Merr.). International Journal of Molecular Sciences. 2019; 20(9):2193. https://doi.org/10.3390/ijms20092193
Chicago/Turabian StyleAl Amin, G M, Keke Kong, Ripa Akter Sharmin, Jiejie Kong, Javaid Akhter Bhat, and Tuanjie Zhao. 2019. "Characterization and Rapid Gene-Mapping of Leaf Lesion Mimic Phenotype of spl-1 Mutant in Soybean (Glycine max (L.) Merr.)" International Journal of Molecular Sciences 20, no. 9: 2193. https://doi.org/10.3390/ijms20092193
APA StyleAl Amin, G. M., Kong, K., Sharmin, R. A., Kong, J., Bhat, J. A., & Zhao, T. (2019). Characterization and Rapid Gene-Mapping of Leaf Lesion Mimic Phenotype of spl-1 Mutant in Soybean (Glycine max (L.) Merr.). International Journal of Molecular Sciences, 20(9), 2193. https://doi.org/10.3390/ijms20092193