Methods for the Refinement of Protein Structure 3D Models


Abstract
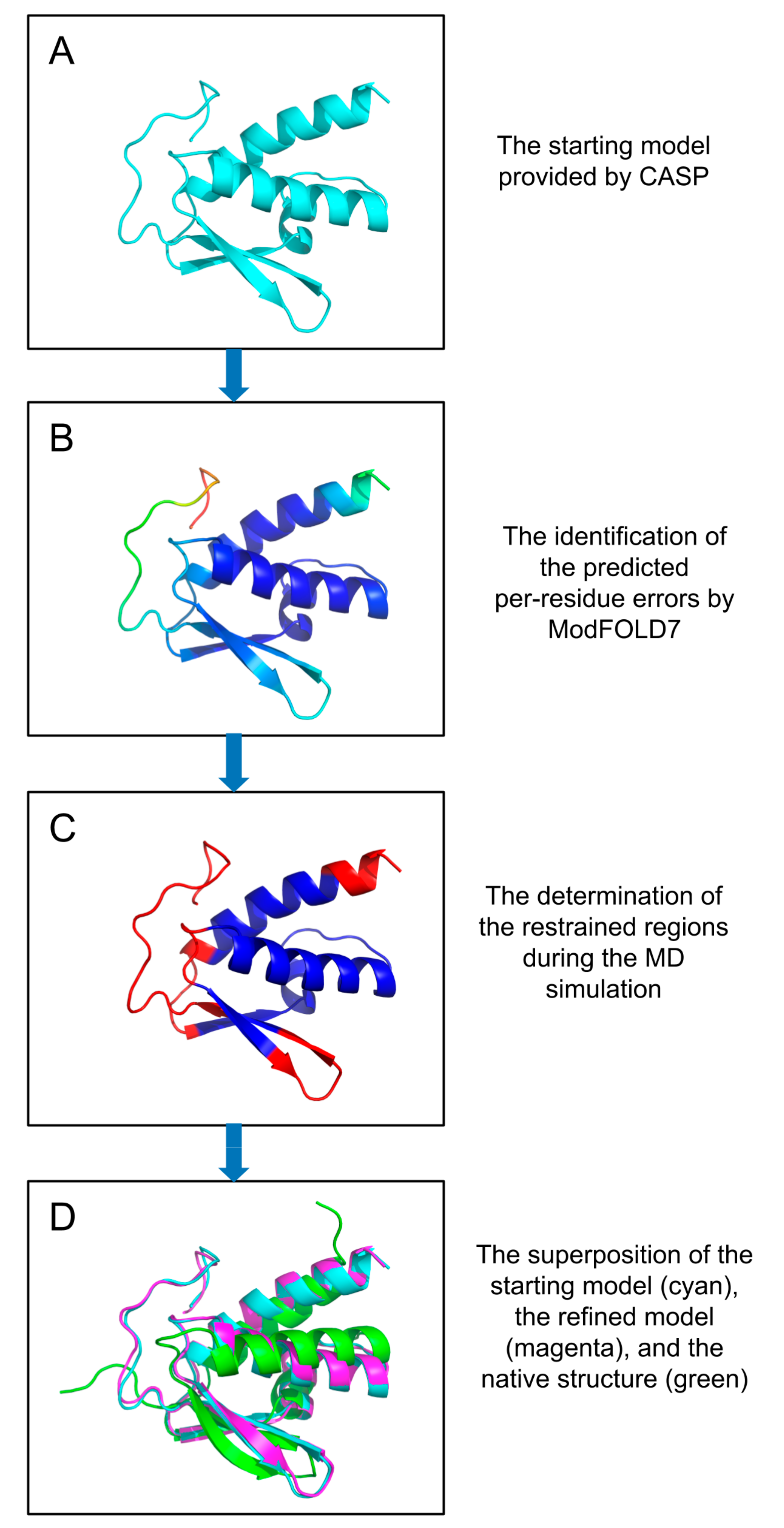
: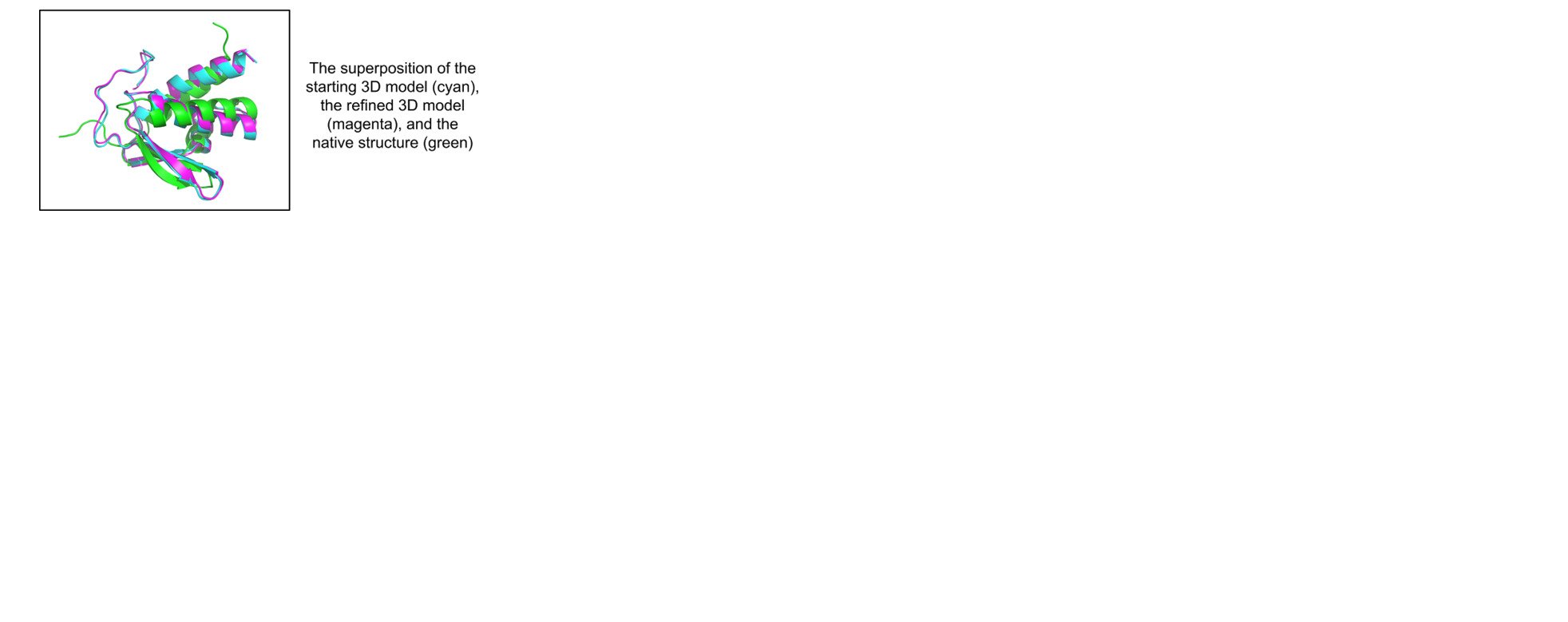
1. Introduction
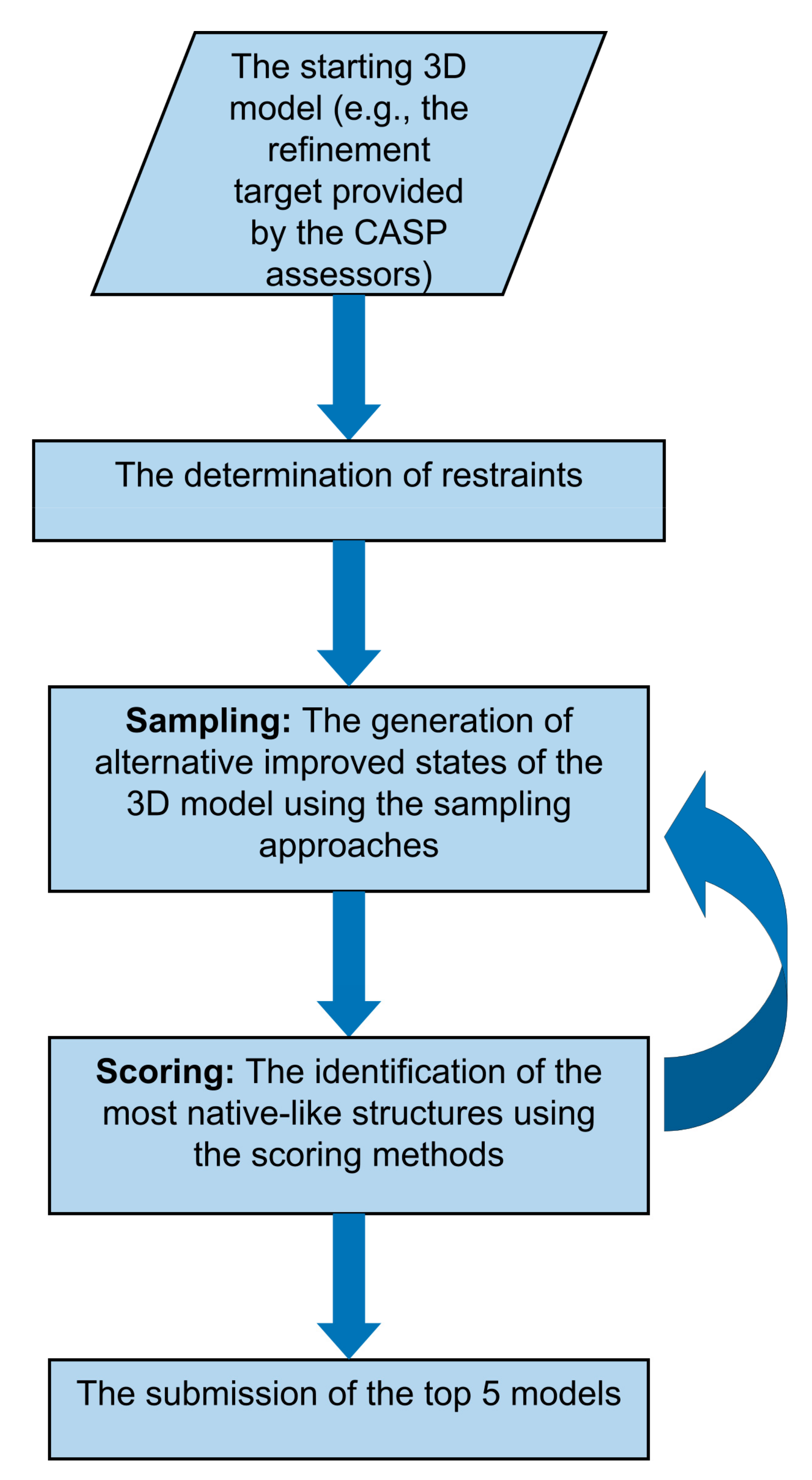
2. Sampling Strategies
Sampling Protocols
3. Scoring Strategies
4. CASP: The Critical Assessment of Techniques for Protein Structure Prediction
4.1. The Refinement Category in CASP Experiments
4.2. Progress with Refinement Strategies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NMR | Nuclear Magnetic Resonance |
| CPU | Central Processing Unit |
| GPU | graphics processing unit |
| Cryo-EM | cryo-electron microscopy |
| PDB | Protein Data Bank |
| CASP | Critical Assessment of techniques for Structure Prediction |
| CHARMM | Chemistry at Harvard Macromolecular Mechanics |
| SVM | Support Vector Machine |
| NAMD | Nanoscale Molecular Dynamics |
| LDDT | Local Distance Difference Test on All Atoms |
| TBM | Template-Based Modelling |
| FM | Free Modelling |
| MQAPs | Model Quality Assessment Programs |
| MD | Molecular Dynamics |
| DFIRE | Distance-Scaled, Finite-Ideal Gas Reference |
| DDFIRE | Dipolar Distance-Scaled, Ideal Gas Reference |
| RWplus | Random Walk reference state Plus |
| GDT-TS | Global Distance Test Total Score |
| GDT_HA | Global Distance Test High Accuracy |
| SphGr | SphereGrinder |
| RMSD | Root mean square deviation |
| TM-Score | Template Modeling Score |
References
- McGuffin, L.J. Aligning Sequences to Structures. In Protein Structure Prediction; Humana Press: Totowa, NJ, USA, 2008; pp. 61–90. [Google Scholar]
- McGuffin, L.J. Protein Fold Recognition and Threading. In Computational Structural Biology; WORLD SCIENTIFIC: Singapore, 2008; pp. 37–60. [Google Scholar]
- Perutz, M.F.; Rossmann, M.G.; Cullis, A.F.; Muirhead, H.; Will, G.; North, A.C.T. Structure of Hæmoglobin: A Three-Dimensional Fourier Synthesis at 5.5-Å. Resolution, Obtained by X-Ray Analysis. Nature 1960, 185, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Kendrew, J.C.; Bodo, G.; Dintzis, H.M.; Parrish, R.G.; Wyckoff, H.; Phillips, D.C. A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis. Nature 1958, 181, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Feig, M. Computational protein structure refinement: Almost there, yet still so far to go. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1307. [Google Scholar] [CrossRef]
- Petsko, G.A.; Ringe, D. Protein Structure and Function; New Science Press: London, UK, 2004; ISBN 9781405119221. [Google Scholar]
- Drenth, J. Principles of Protein X-Ray Crystallography. Springer: Berlin/Heidelberg, Germany, 1999; ISBN 0387985875. [Google Scholar]
- Heinemann, U.; Frevert, J.; Hofman, K.-P.; Illing, G.; Oschkinat, H.; Saenger, W.; Zettl, R. Linking Structural Biology With Genome Research. In Genomics and Proteomics; Kluwer Academic Publishers: Boston, MA, USA, 2002; pp. 179–189. [Google Scholar]
- Murata, K.; Wolf, M. Cryo-electron microscopy for structural analysis of dynamic biological macromolecules. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Jonic, S.; Vénien-Bryan, C. Protein structure determination by electron cryo-microscopy. Curr. Opin. Pharmacol. 2009, 9, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Brocchieri, L.; Karlin, S. Protein length in eukaryotic and prokaryotic proteomes. Nucleic Acids Res. 2005, 33, 3390–3400. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, H.; Karypis, G. Introduction to Protein Structure Prediction: Methods and Algorithms; Wiley: New York, NY, USA, 2010; ISBN 9780470470596. [Google Scholar]
- Roche, D.B.; Buenavista, M.T.; McGuffin, L.J. Protein Structure Prediction and Structural Annotation of Proteomes. In Encyclopedia of Biophysics; Roberts, G.C.K., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 2061–2068. [Google Scholar]
- Stoker, H.S. Organic and Biological Chemistry, 6th ed.; White, A., Ed.; Cengage Learning, Brooks/Cole: Boston, MA, USA, 2013; ISBN 1133103952. [Google Scholar]
- Roche, D.B.; Buenavista, M.T.; McGuffin, L.J. FunFOLDQA: A Quality Assessment Tool for Protein-Ligand Binding Site Residue Predictions. PloS ONE 2012, 7, e38219. [Google Scholar] [CrossRef] [PubMed]
- Pavlopoulou, A.; Michalopoulos, I. State-of-the-art bioinformatics protein structure prediction tools (Review). Int. J. Mol. Med. 2011, 28, 295–310. [Google Scholar]
- Moult, J.; Fidelis, K.; Zemla, A.; Hubbard, T. Critical assessment of methods of protein structure prediction (CASP)-round V. Proteins Struct. Funct. Genet. 2003, 53, 334–339. [Google Scholar] [CrossRef]
- Bradley, P.; Misura, K.M.S.; Baker, D. Toward High-Resolution de Novo Structure Prediction for Small Proteins. Science 2005, 309, 1868–1871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, S.; Zhu, Q.; Zhou, Y. A Knowledge-Based Energy Function for Protein–Ligand, Protein–Protein, and Protein–DNA Complexes. J. Med. Chem. 2005, 48, 2325–2335. [Google Scholar] [CrossRef]
- Ginalski, K.; Grishin, N.V.; Godzik, A.; Rychlewski, L. Practical lessons from protein structure prediction. Nucleic Acids Res. 2005, 33, 1874–1891. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Wu, S.; Zhang, Y. Ab Initio Protein Structure Prediction. In From Protein Structure to Function with Bioinformatics; Springer: Dordrecht, The Netherlands, 2009; pp. 3–25. [Google Scholar]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Rost, B.; Hubbard, T.; Tramontano, A. Critical assessment of methods of protein structure prediction—Round VII. Proteins Struct. Funct. Bioinform. 2007, 69, 3–9. [Google Scholar] [CrossRef]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical assessment of methods of protein structure prediction: Progress and new directions in round XI. Proteins Struct. Funct. Bioinform. 2016, 84, 4–14. [Google Scholar] [CrossRef]
- Moult, J.; Fidelis, K.; Rost, B.; Hubbard, T.; Tramontano, A. Critical assessment of methods of protein structure prediction (CASP)—Round 6. Proteins Struct. Funct. Bioinform. 2005, 61, 3–7. [Google Scholar] [CrossRef]
- Moult, J.; Pedersen, J.T.; Judson, R.; Fidelis, K. A large-scale experiment to assess protein structure prediction methods. Proteins Struct. Funct. Genet. 1995, 23, ii–iv. [Google Scholar] [CrossRef]
- Moult, J. A decade of CASP: Progress, bottlenecks and prognosis in protein structure prediction. Curr. Opin. Struct. Biol. 2005, 15, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Tramontano, A.; Morea, V. Assessment of homology-based predictions in CASP5. Proteins Struct. Funct. Genet. 2003, 53, 352–368. [Google Scholar] [CrossRef] [PubMed]
- Lance, B.K.; Deane, C.M.; Wood, G.R. Exploring the potential of template-based modelling. Bioinformatics 2010, 26, 1849–1856. [Google Scholar] [CrossRef]
- Joo, K.; Lee, J.; Lee, S.; Seo, J.-H.; Lee, S.J.; Lee, J. High accuracy template based modeling by global optimization. Proteins Struct. Funct. Bioinforma. 2007, 69, 83–89. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Mirjalili, V.; Noyes, K.; Feig, M. Physics-based protein structure refinement through multiple molecular dynamics trajectories and structure averaging. Proteins Struct. Funct. Bioinform. 2014, 82, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.B.; Meyer, E.F.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank. A Computer-Based Archival File for Macromolecular Structures. Eur. J. Biochem. 1977, 80, 319–324. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Fischer, D. 3D-SHOTGUN: A novel, cooperative, fold-recognition meta-predictor. Proteins Struct. Funct. Genet. 2003, 51, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Montelione, G.T. Structural genomics: An approach to the protein folding problem. Proc. Natl. Acad. Sci. USA 2001, 98, 13488–13489. [Google Scholar] [CrossRef]
- Westbrook, J.; Feng, Z.; Chen, L.; Yang, H.; Berman, H.M. The Protein Data Bank and structural genomics. Nucleic Acids Res. 2003, 31, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, M.; Edwards, A.; Arrowsmith, C.H.; Montelione, G.T. Structural genomics: Current progress. Science 2003, 299, 1663. [Google Scholar] [CrossRef]
- Baker, D.; Sali, A. Protein structure prediction and structural genomics. Science 2001, 294, 93–96. [Google Scholar] [CrossRef]
- Roche, D.B.; Buenavista, M.T.; Tetchner, S.J.; McGuffin, L.J. The IntFOLD server: An integrated web resource for protein fold recognition, 3D model quality assessment, intrinsic disorder prediction, domain prediction and ligand binding site prediction. Nucleic Acids Res. 2011, 39, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Cheng, J. 3Drefine: Consistent protein structure refinement by optimizing hydrogen bonding network and atomic-level energy minimization. Proteins 2013, 81, 119–131. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Buenavista, M.T.; Roche, D.B. The ModFOLD4 server for the quality assessment of 3D protein models. Nucleic Acids Res. 2013, 41, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Shuid, A.N.; Kempster, R.; McGuffin, L.J. ReFOLD: A server for the refinement of 3D protein models guided by accurate quality estimates. Nucleic Acids Res. 2017, 45, W422–W428. [Google Scholar] [CrossRef] [PubMed]
- Brylinski, M.; Skolnick, J. A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc. Natl. Acad. Sci. USA 2008, 105, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, R.; Tsai, J.; Ruczinski, I.; Baker, D. Functional Inferences from Blind ab Initio Protein Structure Predictions. J. Struct. Biol. 2001, 134, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Wieman, H.; Tøndel, K.; Anderssen, E.; Drabløs, F. Homology-based modelling of targets for rational drug design. Mini Rev. Med. Chem. 2004, 4, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Protein structure prediction: When is it useful? Curr. Opin. Struct. Biol. 2009, 19, 145–155. [Google Scholar] [CrossRef]
- Mirjalili, V.; Feig, M. Protein Structure Refinement through Structure Selection and Averaging from Molecular Dynamics Ensembles. J. Chem. Theory Comput. 2013, 9, 1294–1303. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Watson, J.D.; Thornton, J.M. ProFunc: A server for predicting protein function from 3D structure. Nucleic Acids Res. 2005, 33, W89–W93. [Google Scholar] [CrossRef]
- Becker, O.M.; Dhanoa, D.S.; Marantz, Y.; Chen, D.; Shacham, S.; Cheruku, S.; Heifetz, A.; Mohanty, P.; Fichman, M.; Sharadendu, A.; et al. An Integrated in Silico 3D Model-Driven Discovery of a Novel, Potent, and Selective Amidosulfonamide 5-HT1A Agonist (PRX-00023) for the Treatment of Anxiety and Depression. J. Med. Chem. 2006, 49, 3116–3135. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Applications to targets and beyond. Br. J. Pharmacol. 2007, 152, 21–37. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Cheng, J. i3Drefine Software for Protein 3D Structure Refinement and Its Assessment in CASP10. PloS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Hovan, L.; Oleinikovas, V.; Yalinca, H.; Kryshtafovych, A.; Saladino, G.; Gervasio, F.L. Assessment of the model refinement category in CASP12. Proteins Struct. Funct. Bioinforma. 2018, 86, 152–167. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Feig, M. What makes it difficult to refine protein models further via molecular dynamics simulations? Proteins Struct. Funct. Bioinform. 2018, 86, 177–188. [Google Scholar] [CrossRef]
- Khoury, G.A.; Smadbeck, J.; Kieslich, C.A.; Koskosidis, A.J.; Guzman, Y.A.; Tamamis, P.; Floudas, C.A. Princeton_TIGRESS 2.0: High refinement consistency and net gains through support vector machines and molecular dynamics in double-blind predictions during the CASP11 experiment. Proteins Struct. Funct. Bioinform. 2017, 85, 1078–1098. [Google Scholar] [CrossRef] [PubMed]
- MacCallum, J.L.; Hua, L.; Schnieders, M.J.; Pande, V.S.; Jacobson, M.P.; Dill, K.A. Assessment of the protein-structure refinement category in CASP8. Proteins Struct. Funct. Bioinform. 2009, 77, 66–80. [Google Scholar] [CrossRef]
- MacCallum, J.L.; Pérez, A.; Schnieders, M.J.; Hua, L.; Jacobson, M.P.; Dill, K.A. Assessment of protein structure refinement in CASP9. Proteins Struct. Funct. Bioinform. 2011, 79, 74–90. [Google Scholar] [CrossRef]
- Terashi, G.; Kihara, D. Protein structure model refinement in CASP12 using short and long molecular dynamics simulations in implicit solvent. Proteins Struct. Funct. Bioinform. 2018, 86, 189–201. [Google Scholar] [CrossRef]
- Meiler, J.; Baker, D. Rapid protein fold determination using unassigned NMR data. Proc. Natl. Acad. Sci. USA 2003, 100, 15404–15409. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, A.; Raimondo, D.; Miele, A.E.; Tramontano, A. Evaluating the usefulness of protein structure models for molecular replacement. Bioinformatics 2005, 21, ii72–ii76. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.; Raman, S.; Das, R.; Bradley, P.; McCoy, A.J.; Read, R.J.; Baker, D. High-resolution structure prediction and the crystallographic phase problem. Nature 2007, 450, 259–264. [Google Scholar] [CrossRef]
- Nugent, T.; Cozzetto, D.; Jones, D.T. Evaluation of predictions in the CASP10 model refinement category. Proteins Struct. Funct. Bioinform. 2014, 82, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Modi, V.; Dunbrack, R.L. Assessment of refinement of template-based models in CASP11. Proteins 2016, 260–281. [Google Scholar] [CrossRef]
- Rodrigues, J.P.G.L.M.; Levitt, M.; Chopra, G. KoBaMIN: A knowledge-based minimization web server for protein structure refinement. Nucleic Acids Res. 2012, 40, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Misura, K.M.S.S.; Baker, D. Progress and challenges in high-resolution refinement of protein structure models. Proteins Struct. Funct. Genet. 2005, 59, 15–29. [Google Scholar] [CrossRef]
- Jagielska, A.; Wroblewska, L.; Skolnick, J. Protein model refinement using an optimized physics-based all-atom force field. Proc. Natl. Acad. Sci. USA 2008, 105, 8268–8273. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.S.; Head-Gordon, T. Reliable protein structure refinement using a physical energy function. J. Comput. Chem. 2011, 32, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Skolnick, J. Application of statistical potentials to protein structure refinement from low resolutionab initio models. Biopolymers 2003, 70, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Chopra, G.; Kalisman, N.; Levitt, M. Consistent refinement of submitted models at CASP using a knowledge-based potential. Proteins Struct. Funct. Bioinform. 2010, 78, 2668–2678. [Google Scholar] [CrossRef]
- Han, R.; Leo-Macias, A.; Zerbino, D.; Bastolla, U.; Contreras-Moreira, B.; Ortiz, A.R. An efficient conformational sampling method for homology modeling. Proteins Struct. Funct. Bioinform. 2008, 71, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Blum, B.; Bradley, P.; Baker, D. Sampling Bottlenecks in De novo Protein Structure Prediction. J. Mol. Biol. 2009, 393, 249–260. [Google Scholar] [CrossRef]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.W.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods Enzymol. 2011, 487, 545–574. [Google Scholar]
- Song, Y.; DiMaio, F.; Wang, R.Y.-R.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-Resolution Comparative Modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, S.; Park, H.; Kim, D.E.; DiMaio, F.; Baker, D. Protein structure prediction using Rosetta in CASP12. Proteins Struct. Funct. Bioinform. 2018, 86, 113–121. [Google Scholar] [CrossRef]
- Summa, C.M.; Levitt, M. Near-native structure refinement using in vacuo energy minimization. Proc. Natl. Acad. Sci. USA 2007, 104, 3177–3182. [Google Scholar] [CrossRef]
- Fan, H.; Mark, A.E. Refinement of homology-based protein structures by molecular dynamics simulation techniques. Protein Sci. 2004, 13, 211–220. [Google Scholar] [CrossRef]
- Chen, J.; Brooks, C.L. Can molecular dynamics simulations provide high-resolution refinement of protein structure? Proteins Struct. Funct. Bioinform. 2007, 67, 922–930. [Google Scholar] [CrossRef]
- Ishitani, R.; Terada, T.; Shimizu, K. Refinement of comparative models of protein structure by using multicanonical molecular dynamics simulations. Mol. Simul. 2008, 34, 327–336. [Google Scholar] [CrossRef]
- Kannan, S.; Zacharias, M. Application of biasing-potential replica-exchange simulations for loop modeling and refinement of proteins in explicit solvent. Proteins Struct. Funct. Bioinform. 2010, 78, 2809–2819. [Google Scholar] [CrossRef]
- Gront, D.; Kmiecik, S.; Blaszczyk, M.; Ekonomiuk, D.; Koliński, A. Optimization of protein models. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 479–493. [Google Scholar] [CrossRef]
- Chen, J.; Brooks, C.L.; Khandogin, J. Recent advances in implicit solvent-based methods for biomolecular simulations. Curr. Opin. Struct. Biol. 2008, 18, 140–148. [Google Scholar] [CrossRef]
- Heo, L.; Feig, M. PREFMD: A web server for protein structure refinement via molecular dynamics simulations. Bioinformatics 2018, 34, 1063–1065. [Google Scholar] [CrossRef]
- Feig, M. Local Protein Structure Refinement via Molecular Dynamics Simulations with locPREFMD. J. Chem. Inf. Model. 2016, 56, 1304–1312. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Lee, G.R.; Heo, L.; Seok, C. Effective protein model structure refinement by loop modeling and overall relaxation. Proteins Struct. Funct. Bioinform. 2016, 84, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Tamamis, P.; Pinnaduwage, N.; Smadbeck, J.; Kieslich, C.A.; Floudas, C.A. Princeton_TIGRESS: Protein geometry refinement using simulations and support vector machines. Proteins Struct. Funct. Bioinform. 2014, 82, 794–814. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.; Piana, S.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Refinement of protein structure homology models via long, all-atom molecular dynamics simulations. Proteins Struct. Funct. Bioinform. 2012, 80, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How Fast-Folding Proteins Fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Fan, H.; Periole, X.; Honig, B.; Mark, A.E. Refining homology models by combining replica-exchange molecular dynamics and statistical potentials. Proteins Struct. Funct. Bioinform. 2008, 72, 1171–1188. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.R.; Tsai, J.; Baker, D.; Kollman, P.A. Molecular dynamics in the endgame of protein structure prediction. J. Mol. Biol. 2001, 313, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Feig, M. Experimental accuracy in protein structure refinement via molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2018, 115, 13276–13281. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Systematic Validation of Protein Force Fields against Experimental Data. PloS ONE 2012, 7, e32131. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ 1 and χ 2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Buchete, N.-V.; Hummer, G. Are Current Molecular Dynamics Force Fields too Helical? Biophys. J. 2008, 95, L07–L09. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef]
- MacPherson, S.; Larochelle, M.; Turcotte, B.; Link, C.; Kann, M.; Swinkels, J.W.; Kornegay, E.T.; Verstegen, M.W.; Tertiary, P.; Structures, Q.; et al. Computational Protein Design: A problem in combinatorial optimization. What is a protein? Nutr. Res. Rev. 2004, 11, 205–229. [Google Scholar]
- MacKerell, A.D.; Banavali, N.; Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 2001, 56, 257–265. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields. J. Comp. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, S.; Kim, D.E.; Wang, R.Y.-R.; Liu, Y.; DiMaio, F.; Baker, D. Improved de novo structure prediction in CASP11 by incorporating coevolution information into Rosetta. Proteins Struct. Funct. Bioinforma. 2016, 84, 67–75. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.; Cheatham, T.E.; Wang, J.; Duck, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; Roberts, B.P.; et al. Amber 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Cheng, Q.; Joung, I.; Lee, J. A Simple and Efficient Protein Structure Refinement Method. J. Chem. Theory Comput. 2017, 13, 5146–5162. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Gangupomu, V.; Wagner, J.; Jain, A.; Vaidehi, N. Structure Refinement of Protein Low Resolution Models Using the GNEIMO Constrained Dynamics Method. J. Phys. Chem. B 2012, 116, 2365–2375. [Google Scholar] [CrossRef]
- Feig, M.; Mirjalili, V. Protein Structure Refinement via Molecular-Dynamics Simulations: What works and what does not? Proteins Struct. Funct. Bioinforma. 2016, 84, 282–292. [Google Scholar] [CrossRef]
- Cao, W.; Terada, T.; Nakamura, S.; Shimizu, K. Refinement of Comparative-Modeling Structures by Multicanonical Molecular Dynamics. Genome Inform. 2003, 14, 484–485. [Google Scholar]
- Zhang, J.; Liang, Y.; Zhang, Y. Atomic-Level Protein Structure Refinement Using Fragment-Guided Molecular Dynamics Conformation Sampling. Structure 2011, 19, 1784–1795. [Google Scholar] [CrossRef]
- Park, H.; Seok, C. Refinement of Unreliable Local Regions in Template-based Protein Models. Proteins Struct. Funct. Bioinform. 2012, 80, 1974–1986. [Google Scholar] [CrossRef]
- Maghrabi, A.H.A.; McGuffin, L.J. ModFOLD6: An accurate web server for the global and local quality estimation of 3D protein models. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Critical Assessment of Techniques for Protein Structure Prediction. 13 Abstracts. Available online: http://predictioncenter.org/casp13/index.cgi (accessed on 2 April 2019).
- Seemayer, S.; Gruber, M.; Söding, J. CCMpred—Fast and precise prediction of protein residue-residue contacts from correlated mutations. Bioinformatics 2014, 30, 3128–3130. [Google Scholar] [CrossRef]
- Liu, Y.; Palmedo, P.; Ye, Q.; Berger, B.; Peng, J. Enhancing Evolutionary Couplings with Deep Convolutional Neural Networks. Cell Syst. 2018, 6, 65.e3–74.e3. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Lee, H.; Seok, C. GalaxyRefineComplex: Refinement of protein-protein complex model structures driven by interface repacking. Sci. Rep. 2016, 6, 32153. [Google Scholar] [CrossRef]
- Güntert, P. Automated NMR Structure Calculation With CYANA. In Protein NMR Techniques; Humana Press: Totowa, NJ, USA, 2004; Volume 278, pp. 353–378. [Google Scholar]
- Yang, Y.; Zhou, Y. Specific interactions for ab initio folding of protein terminal regions with secondary structures. Proteins Struct. Funct. Bioinform. 2008, 72, 793–803. [Google Scholar] [CrossRef]
- Cossio, P.; Granata, D.; Laio, A.; Seno, F.; Trovato, A. A simple and efficient statistical potential for scoring ensembles of protein structures. Sci. Rep. 2012, 2, 351. [Google Scholar] [CrossRef]
- Kuhlman, B.; Dantas, G.; Ireton, G.C.; Varani, G.; Stoddard, B.L.; Baker, D. Design of a novel globular protein fold with atomic-level accuracy. Science 2003, 302, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Kalisman, N.; Levi, A.; Maximova, T.; Reshef, D.; Zafriri-Lynn, S.; Gleyzer, Y.; Keasar, C. MESHI: A new library of Java classes for molecular modeling. Bioinformatics 2005, 21, 3931–3932. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Atkins, J.D.; Salehe, B.R.; Shuid, A.N.; Roche, D.B. IntFOLD: An integrated server for modelling protein structures and functions from amino acid sequences. Nucleic Acids Res. 2015, 43, W169–W173. [Google Scholar] [CrossRef] [PubMed]
- Rykunov, D.; Fiser, A. New statistical potential for quality assessment of protein models and a survey of energy functions. BMC Bioinform. 2010, 11, 128. [Google Scholar] [CrossRef]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; DiMaio, F.; Baker, D. The Origin of Consistent Protein Structure Refinement from Structural Averaging. Structure 2015, 23, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Stumpff-Kane, A.W.; Maksimiak, K.; Lee, M.S.; Feig, M. Sampling of near-native protein conformations during protein structure refinement using a coarse-grained model, normal modes, and molecular dynamics simulations. Proteins Struct. Funct. Bioinform. 2007, 70, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.B.; Wagner, J.R.; Jain, A.; Vaidehi, N. Protein Structure Refinement of CASP Target Proteins Using GNEIMO Torsional Dynamics Method. J. Chem. Inf. Model. 2014, 54, 508–517. [Google Scholar] [CrossRef]
- Olson, M.A.; Lee, M.S. Evaluation of Unrestrained Replica-Exchange Simulations Using Dynamic Walkers in Temperature Space for Protein Structure Refinement. PloS ONE 2014, 9, e96638. [Google Scholar] [CrossRef]
- Kumar, A.; Campitelli, P.; Thorpe, M.F.; Ozkan, S.B. Partial unfolding and refolding for structure refinement: A unified approach of geometric simulations and molecular dynamics. Proteins Struct. Funct. Bioinform. 2015, 83, 2279–2292. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. SPICKER: A clustering approach to identify near-native protein folds. J. Comput. Chem. 2004, 25, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins Struct. Funct. Bioinform. 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y. A Novel Side-Chain Orientation Dependent Potential Derived from Random-Walk Reference State for Protein Fold Selection and Structure Prediction. PloS ONE 2010, 5, e15386. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef]
- Lu, M.; Dousis, A.D.; Ma, J. OPUS-PSP: An orientation-dependent statistical all-atom potential derived from side-chain packing. J. Mol. Biol. 2008, 376, 288–301. [Google Scholar] [CrossRef]
- Tyka, M.D.; Keedy, D.A.; André, I.; Dimaio, F.; Song, Y.; Richardson, D.C.; Richardson, J.S.; Baker, D. Alternate states of proteins revealed by detailed energy landscape mapping. J. Mol. Biol. 2011, 405, 607–618. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, F.; Tyka, M.D.; Baker, M.L.; Chiu, W.; Baker, D. Refinement of Protein Structures into Low-Resolution Density Maps Using Rosetta. J. Mol. Biol. 2009, 392, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Klebe, G. Statistical potentials and scoring functions applied to protein–ligand binding. Curr. Opin. Struct. Biol. 2001, 11, 231–235. [Google Scholar] [CrossRef]
- Russ, W.P.; Ranganathan, R. Knowledge-based potential functions in protein design. Curr. Opin. Struct. Biol. 2002, 12, 447–452. [Google Scholar] [CrossRef]
- Buchete, N.-V.; Straub, J.; Thirumalai, D. Development of novel statistical potentials for protein fold recognition. Curr. Opin. Struct. Biol. 2004, 14, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, H.; Zhang, C.; Liu, S. What is a Desirable Statistical Energy Function for Proteins and How Can It Be Obtained? Cell Biochem. Biophys. 2006, 46, 165–174. [Google Scholar] [CrossRef]
- Skolnick, J. In quest of an empirical potential for protein structure prediction. Curr. Opin. Struct. Biol. 2006, 16, 166–171. [Google Scholar] [CrossRef]
- Bradley, P.; Malmström, L.; Qian, B.; Schonbrun, J.; Chivian, D.; Kim, D.E.; Meiler, J.; Misura, K.M.S.; Baker, D. Free modeling with Rosetta in CASP6. Proteins Struct. Funct. Bioinform. 2005, 61, 128–134. [Google Scholar] [CrossRef]
- Sippl, M.J. Knowledge-based potentials for proteins. Curr. Opin. Struct. Biol. 1995, 5, 229–235. [Google Scholar] [CrossRef]
- Jernigan, R.L.; Bahar, I. Structure-derived potentials and protein simulations. Curr. Opin. Struct. Biol. 1996, 6, 195–209. [Google Scholar] [CrossRef]
- Moult, J. Comparison of database potentials and molecular mechanics force fields. Curr. Opin. Struct. Biol. 1997, 7, 194–199. [Google Scholar] [CrossRef]
- Lazaridis, T.; Karplus, M. Effective energy functions for protein structure prediction. Curr. Opin. Struct. Biol. 2000, 10, 139–145. [Google Scholar] [CrossRef]
- Dutagaci, B.; Heo, L.; Feig, M. Structure refinement of membrane proteins via molecular dynamics simulations. Proteins Struct. Funct. Bioinform. 2018, 86, 738–750. [Google Scholar] [CrossRef]
- Olson, M.A.; Lee, M.S. Application of replica exchange umbrella sampling to protein structure refinement of nontemplate models. J. Comput. Chem. 2013, 34, 1785–1793. [Google Scholar] [CrossRef]
- Zhou, H.; Zhou, Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002, 11, 2714–2726. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, Y. Ab initio folding of terminal segments with secondary structures reveals the fine difference between two closely related all-atom statistical energy functions. Protein Sci. 2008, 17, 1212–1219. [Google Scholar] [CrossRef]
- Leaver-Fay, A.; O’Meara, M.J.; Tyka, M.; Jacak, R.; Song, Y.; Kellogg, E.H.; Thompson, J.; Davis, I.W.; Pache, R.A.; Lyskov, S.; et al. Scientific Benchmarks for Guiding Macromolecular Energy Function Improvement. Methods Enzymol. 2013, 523, 109–143. [Google Scholar] [PubMed]
- Rohl, C.A.; Strauss, C.E.M.; Misura, K.M.S.; Baker, D. Protein Structure Prediction Using Rosetta. Methods Enzymol. 2004, 383, 66–93. [Google Scholar]
- Kuhlman, B.; Baker, D. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. USA 2000, 97, 10383–10388. [Google Scholar] [CrossRef]
- Park, H.; DiMaio, F.; Baker, D. CASP11 refinement experiments with ROSETTA. Proteins Struct. Funct. Bioinform. 2016, 84, 314–322. [Google Scholar] [CrossRef]
- Park, H.; Ovchinnikov, S.; Kim, D.E.; DiMaio, F.; Baker, D. Protein homology model refinement by large-scale energy optimization. Proc. Natl. Acad. Sci. USA 2018, 115, 3054–3059. [Google Scholar] [CrossRef] [PubMed]
- Wallner, B.; Elofsson, A. Can correct protein models be identified? Protein Sci. 2003, 12, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Lindahl, E.; Orn Wallner, B. Improved model quality assessment using ProQ2. BMC Bioinform. 2012, 13, 1–12. [Google Scholar] [CrossRef]
- Randall, A.; Baldi, P. SELECTpro: Effective protein model selection using a structure-based energy function resistant to BLUNDERs. BMC Struct. Biol. 2008, 8, 52. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Barbato, A.; Fidelis, K.; Monastyrskyy, B.; Schwede, T.; Tramontano, A. Assessment of the assessment: Evaluation of the model quality estimates in CASP10. Proteins 2014, 82 Suppl. 2, 112–126. [Google Scholar] [CrossRef]
- Larsson, P.; Skwark, M.J.; Wallner, B.; Elofsson, A. Assessment of global and local model quality in CASP8 using Pcons and ProQ. Proteins Struct. Funct. Bioinform. 2009, 77, 167–172. [Google Scholar] [CrossRef]
- Cozzetto, D.; Kryshtafovych, A.; Tramontano, A. Evaluation of CASP8 model quality predictions. Proteins Struct. Funct. Bioinform. 2009, 77, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Venclovas, Č.; Fidelis, K.; Moult, J. Progress over the first decade of CASP experiments. Proteins Struct. Funct. Bioinform. 2005, 61, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Rost, B.; Tramontano, A. Critical assessment of methods of protein structure prediction-Round VIII. Proteins Struct. Funct. Bioinform. 2009, 77, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical assessment of methods of protein structure prediction (CASP)—Round x. Proteins Struct. Funct. Bioinform. 2014, 82, 1–6. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Zemla, A. LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003, 31, 3370–3374. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Monastyrskyy, B.; Fidelis, K. CASP11 statistics and the prediction center evaluation system. Proteins Struct. Funct. Bioinform. 2016, 84, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Monastyrskyy, B.; Fidelis, K. CASP prediction center infrastructure and evaluation measures in CASP10 and CASP ROLL. Proteins Struct. Funct. Bioinform. 2014, 82, 7–13. [Google Scholar] [CrossRef]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef]
- Olechnovič, K.; Kulberkytė, E.; Venclovas, Č. CAD-score: A new contact area difference-based function for evaluation of protein structural models. Proteins Struct. Funct. Bioinform. 2013, 81, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Cong, Q.; Kinch, L.N.; Pei, J.; Shi, S.; Grishin, V.N.; Li, W.; Grishin, N.V. An automatic method for CASP9 free modeling structure prediction assessment. Bioinformatics 2011, 27, 3371–3378. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, B.F.; Stock, A.M.; Ringe, D.; Petsko, G.A. Crystalline ribonuclease A loses function below the dynamical transition at 220 K. Nature 1992, 357, 423–424. [Google Scholar] [CrossRef]
- Eisenmesser, E.Z.; Bosco, D.A.; Akke, M.; Kern, D. Enzyme dynamics during catalysis. Science 2002, 295, 1520–1523. [Google Scholar] [CrossRef]
- Benkovic, S.J.; Hammes-Schiffer, S. A perspective on enzyme catalysis. Science 2003, 301, 1196–1202. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Storoni, L.C.; Read, R.J. IUCr likelihood-enhanced fast translation functions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 458–464. [Google Scholar] [CrossRef]
- Mobley, D.L.; Chodera, J.D.; Dill, K.A. On the use of orientational restraints and symmetry corrections in alchemical free energy calculations. J. Chem. Phys. 2006, 125, 084902. [Google Scholar] [CrossRef]
- Zagrovic, B.; van Gunsteren, W.F. Comparing atomistic simulation data with the NMR experiment: How much can NOEs actually tell us? Proteins Struct. Funct. Bioinform. 2006, 63, 210–218. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Best, R.B.; DePristo, M.A.; Dobson, C.M.; Vendruscolo, M. Simultaneous determination of protein structure and dynamics. Nature 2005, 433, 128–132. [Google Scholar] [CrossRef]
- Senior, A.; Jumper, J.; Hassabis, D. Deep Mind, AlphaFold: Using AI for scientific discovery. Available online: https://deepmind.com/blog/alphafold/ (accessed on 8 May 2019).
- Wallner, B.; Elofsson, A. Prediction of global and local model quality in CASP7 using Pcons and ProQ. Proteins Struct. Funct. Bioinform. 2007, 69, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Uziela, K.; Wallner, B. ProQ2: Estimation of model accuracy implemented in Rosetta. Bioinformatics 2016, 32, 1411–1413. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Name | URL |
|---|---|
| PREFMD [85] | http://feiglab.org/prefmd |
| locPREFMD [86] | http://feig.bch.msu.edu/web/services/locprefmd/ |
| GalaxyRefine [54] | http://galaxy.seoklab.org/refine |
| KoBaMIN [66] | http://csb.stanford.edu/kobamin |
| Princeton_TIGRESS 2.0 [56] | http://atlas.engr.tamu.edu/refinement/ |
| ModRefiner [67] | http://zhanglab.ccmb.med.umich.edu/ModRefiner |
| 3DRefine [41,122] | http://sysbio.rnet.missouri.edu/3Drefine/ |
| ReFOLD [43] | http://www.reading.ac.uk/bioinf/ReFOLD/ |
| FG-MD [110] | http://zhanglab.ccmb.med.umich.edu/FG-MD/ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adiyaman, R.; McGuffin, L.J. Methods for the Refinement of Protein Structure 3D Models. Int. J. Mol. Sci. 2019, 20, 2301. https://doi.org/10.3390/ijms20092301
Adiyaman R, McGuffin LJ. Methods for the Refinement of Protein Structure 3D Models. International Journal of Molecular Sciences. 2019; 20(9):2301. https://doi.org/10.3390/ijms20092301
Chicago/Turabian StyleAdiyaman, Recep, and Liam James McGuffin. 2019. "Methods for the Refinement of Protein Structure 3D Models" International Journal of Molecular Sciences 20, no. 9: 2301. https://doi.org/10.3390/ijms20092301
APA StyleAdiyaman, R., & McGuffin, L. J. (2019). Methods for the Refinement of Protein Structure 3D Models. International Journal of Molecular Sciences, 20(9), 2301. https://doi.org/10.3390/ijms20092301