Inflammatory and Molecular Pathways in Heart Failure—Ischemia, HFpEF and Transthyretin Cardiac Amyloidosis


Abstract
:1. Introduction
2. Pathways in Ischemic Heart Failure
3. Pathways in HFpEF
4. Novel Therapeutics in Heart Failure—Immunosuppression, Immunomodulation, Regeneration
5. Transthyretin Amyloidosis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 6MWT | 6-min walk test |
| 99mTc-DPD | Tc-99m-3,3-diphosphono-1,2-propanodicarboxylicacid |
| AL | light chain amyloidosis |
| APC | antigen presenting cells |
| ASO | anti-sense oligonucleotide |
| ATII | angiotensin II |
| ATTR | transthyretin amyloidosis |
| CCL2 | CC-chemokine ligand 2 |
| CCR2 | C-C chemokine receptor type 2 |
| cGMP | cyclic guanosine monophosphate |
| CMRI | cardiac magnetic resonance imaging |
| CRP | C-reactive protein |
| CsA | cyclosporin A |
| DAMPs | danger-associated molecular patterns |
| DCs | dendritic cells |
| EC | European Commission |
| ECG | electrocardiogram |
| ECM | extracellular matrix |
| FAP | familial amyloid polyneuropathy |
| FDA | Food and Drug Administration |
| hATTR | hereditary transthyretin amyloidosis |
| HF | heart failure |
| HFpEF | heart failure with preserved ejection fraction |
| HFrEF | heart failure with reduced ejection fraction |
| ICAM1 | intracellular adhesion molecule 1 |
| IVIg | immunoglobulins |
| IL-1 | Interleukin-1 |
| IL-1β | Interleukin-1β |
| IL-10 | Interleukin-10 |
| IL-6 | Interleukin-6 |
| LVEF | left ventricular ejection fraction |
| LVH | left ventricular hypertrophy |
| LYVE-1 | lymphatic vessel endothelial hyaluronan receptor 1 |
| MI | myocardial infarction |
| MMPs | matrix metalloproteinases |
| MRI | magnetic resonance imaging |
| MTX | methotrexate |
| NAC | N-acetylcysteine |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B-cells |
| NO | nitric oxide |
| NT-proBNP | N-terminal prohormone of brain natriuretic peptide |
| PCI | percutaneous coronary intervention |
| PKG | protein kinase G |
| ROS | reactive oxygen species |
| SAP | SLAM-associated protein |
| sGC | soluble guanylyl cyclase |
| siRNA | small-interfering RNA |
| STEMI | ST-elevated myocardial infarction |
| TAC | transverse aortic constriction |
| TGF-β | transforming growth factor beta |
| TNF-α | Tumor necrosis factor alpha |
| TNFR1 | TNF receptor 1 |
| TNFR2 | TNF receptor 2 |
| TTR | transthyretin |
| TUDCA | tauroursodeoxycholic acid |
| UDCA | ursodeoxycholic acid |
| VEGF | vascular endothelial growth factor |
| VEGF-C | vascular endothelial growth factor-C |
| VCAM1 | vascular cell adhesion protein 1 |
| wtATTR | wild-type transthyretin amyloidosis |
References
- Ponikowski, P.; Anker, S.D.; AlHabib, K.F.; Cowie, M.R.; Force, T.L.; Hu, S.; Jaarsma, T.; Krum, H.; Rastogi, V.; Rohde, L.E.; et al. Heart failure: Preventing disease and death worldwide. ESC Heart Fail. 2014, 1, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2016, 18, 891–975. [Google Scholar] [PubMed]
- Gladden, J.D.; Linke, W.A.; Redfield, M.M. Heart failure with preserved ejection fraction. Pflugers Arch. 2014, 466, 1037–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantz, S.; Falcao-Pire, S.I.; Balligand, J.L.; Bauersachs, J.; Brutsaert, D.; Ciccarelli, M.; Dawson, D.; de Windt, L.J.; Giacca, M.; Hamdani, N.; et al. The innate immune system in chronic cardiomyopathy: A European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur. Heart Fail. 2018, 20, 445–459. [Google Scholar]
- Lourenҫo, A.P.; Leite-Moreira, A.F.; Balligand, J.L.; Bauersachs, J.; Dawson, D.; de Boer, R.A.; de Windt, L.J.; Falcão-Pires, I.; Fontes-Carvalho, R.; Franz, S.; et al. An integrative translational approach to study heart failure with preserved ejection fraction: A position paper from the Working Group on Myocardial Infarction of the European Society of Cardiology. Eur. J. Heart Fail. 2018, 20, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Van Linthout, S.; Tschöpe, C. Inflammation—Cause or Consequence of Heart Failure or Both? Curr. Heart Fail. Rep. 2017, 14, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tan, G.J.; Han, L.N.; Bai, Y.Y.; He, M.; Liu, H.B. Novel biomarkers for cardiovascular risk prediction. J. Geriatr. Cardiol. 2017, 14, 135–150. [Google Scholar] [PubMed]
- Magnussen, C.; Blankenberg, S. Biomarkers for heart failure: Small molecules with high clinical relevance. J. Intern. Med. 2018, 283, 530–543. [Google Scholar] [CrossRef]
- Piek, A.; Du, W.; de Boer, R.A.; Silljé, H.H.W. Novel heart failure biomarkers: Why do we fail to exploit their potential? Crit. Rev. Clin. Lab. Sci. 2018, 55, 246–263. [Google Scholar] [CrossRef]
- Roh, J.; Houstis, N.; Rosenzweig, A. Why don’t we have proven treatments for HFpEF? Circ. Res. 2017, 120, 1243–1245. [Google Scholar] [CrossRef] [Green Version]
- Savarese, G.; Lund, L.H. Global Public Health Burden of Heart Failure. Card. Fail. Rev. 2017, 3, 7–11. [Google Scholar] [CrossRef] [Green Version]
- González-López, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef] [Green Version]
- Mirzoyev, S.A.; Edwards, W.D.; Mohammed, S.F.; Donovan, J.L.; Roger, V.L.; Grogan, D.R.; Redfield, M.M. Cardiac amyloid deposition is common in elderly patients with heart failure and preserved ejection fraction. Circulation 2010, 122, A17926. [Google Scholar]
- Leuschner, F.; Panizzi, P.; Chico-Calero, I.; Lee, W.W.; Ueno, T.; Cortez-Retamozo, V.; Waterman, P.; Gorbatov, R.; Marinelli, B.; Iwamoto, Y.; Chudnovskiy, A.; et al. Angiotensin-converting enzyme inhibition prevents the release of monocytes from their splenic reservoir in mice with myocardial infarction. Science 2009, 325, 612–616. [Google Scholar] [CrossRef]
- Latet, S.C.; Hoymans, V.Y.; van Herck, P.L.; Vrints, C.J. The cellular immune system in post-myocardial infarction repair process. Int. J. Cardiol. 2015, 179, 240–247. [Google Scholar] [CrossRef]
- Sager, H.B.; Kessler, T.; Schunkert, H. Monocytes and macrophages in cardiac injury and repair. J. Thorac. Dis. 2017, 9, 30–35. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Wissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [Green Version]
- Dewald, O.; Zymek, P.; Winkelmann, K.; Koerting, A.; Ren, G.; Abou-Khamis, T.; Michael, L.H.; Rollins, B.J.; Entman, M.L.; Frangogiannis, N.G. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 2005, 96, 881–889. [Google Scholar] [CrossRef]
- Sager, H.B.; Hulsmans, M.; Lavine, K.J.; Moreira, M.B.; Heidt, T.; Courties, G.; Sun, Y.; Iwamoto, Y.; Tricot, B.; Khan, O.F.; et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ. Res. 2016, 119, 853–864. [Google Scholar] [CrossRef]
- Xuan, W.; Qu, Q.; Zheng, B.; Xiong, S.; Fan, G.H. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J. Leukoc. Biol. 2015, 97, 61–69. [Google Scholar] [CrossRef]
- Monden, Y.; Kubota, T.; Inoue, T.; Tsutsumi, T.; Kawano, S.; Ide, T.; Tsutsui, H.; Sunagawa, K. Tumor necrosis factor-alpha is toxic via receptor 1 and protective via receptor 2 in a murine model of myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, 743–753. [Google Scholar] [CrossRef]
- Schumacher, S.M.; Prasad, S.V.N. Tumor Necrosis Factor-α in Heart Failure: An updated review. Curr. Cardiol. Rep. 2018, 20, 117. [Google Scholar] [CrossRef]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple Facets of NF-κB in the Heart: To Be or Not to NF-κB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef]
- Hulsmans, M.; Sam, F.; Nahrendorf, M. Monocyte and macrophage contributions to cardiac remodeling. J. Mol. Cell Cardiol. 2016, 93, 149–155. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef]
- Kumar, K.P.; Nicholls, A.J.; Wong, C.H.Y. Partners in crime: Neutrophils and monocytes/ macrophages in inflammation and disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef]
- Ben-Mordechai, T.; Palevski, D.; Glucksam-Galnoy, Y.; Elron-Gross, I.; Margalit, R.; Leor, J. Targeting macrophage subsets for infarct repair. J. Cardiovasc. Pharmacol. Ther. 2015, 20, 36–51. [Google Scholar] [CrossRef]
- Frantz, S.; Hofmann, U.; Fraccarollo, D.; Schäfer, A.; Kranepuhl, S.; Hagedorn, I.; Nieswandt, B.; Nahrendorf, M.; Wagner, H.; Bayer, B.; et al. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J. 2013, 27, 871–881. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodeling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Chen, W.; Frangogiannis, N.G. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim. Biophys. Acta 2013, 1833, 945–953. [Google Scholar] [CrossRef]
- Saxena, A.; Chen, W.; Su, Y.; Rai, V.; Uche, O.U.; Li, N.; Frangogiannis, N.G. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J. Immunol. 2013, 191, 4838–4848. [Google Scholar] [CrossRef]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction—From repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Ismahil, M.A.; Hamid, T.; Bansal, S.S.; Patel, B.; Kingery, J.R.; Prabhu, S.D. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: Critical importance of the cardiosplenic axis. Circ. Res. 2014, 114, 266–282. [Google Scholar] [CrossRef]
- Mann, D.L. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef]
- Sager, H.B.; Dutta, P.; Dahlman, J.E.; Hulsmans, M.; Courties, G.; Sun, Y.; Heidt, T.; Vinegoni, C.; Brodovsky, A.; Fitzgerald, K.; et al. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci. Transl. Med. 2016, 8, 342ra80. [Google Scholar] [CrossRef]
- Maekawa, Y.; Anzai, T.; Yoshikawa, T.; Asakura, Y.; Takahashi, T.; Ishikawa, S.; Mitamura, H.; Ogawa, S. Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction: A possible role for left ventricular remodeling. J. Am. Coll. Cardiol. 2002, 39, 241–246. [Google Scholar] [CrossRef]
- Huang, L.H.; Lavine, K.J.; Randolph, G.J. Cardiac Lymphatic Vessels, Transport, and Healing of the Infarcted Heart. JACC Basic Transl. Sci. 2017, 2, 477–483. [Google Scholar] [CrossRef]
- Vieira, J.M.; Norman, S.; Villa Del Campo, C.; Cahill, T.J.; Barnette, D.N.; Gunadasa-Rohling, M.; Johnson, L.A.; Greaves, D.R.; Carr, C.A.; Jackson, D.G.; et al. The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J. Clin. Invest. 2018, 128, 3402–3412. [Google Scholar] [CrossRef]
- Kaya, Z.; Leib, C.; Katus, H.A. Autoantibodies in heart failure and cardiac dysfunction. Circ. Res. 2012, 110, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Anzai, A.; Katsumata, Y.; Matsuhashi, T.; Ito, K.; Endo, J.; Yamamoto, T.; Takeshima, A.; Shinmura, K.; Shen, W.; et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell. Cardiol. 2013, 62, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Lipes, M.A. Role of impaired central tolerance to alpha-myosin in inflammatory heart disease. Trends Cardiovasc. Med. 2012, 22, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Land, W.G. The role of damage-associated molecular patterns (DAMPs) in human disease: Part II: DAMPs as diagnostics, prognostics and therapeutics in clinical medicine. Sultan Qaboos Univ. Med. J. 2015, 15, e157–e170. [Google Scholar] [PubMed]
- Sattler, S.; Fairchild, P.; Watt, F.M.; Rosenthal, N.; Harding, SE. The adaptive immune response to cardiac injury—The true roadblock to effective regenerative therapies? Regen. Med. 2017, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Reyes, A.M.; Youker, K.A.; Trevino, A.R.; Celis, R.; Hamilton, D.J.; Flores-Arredondo, J.H.; Orrego, C.M.; Bhimaraj, A.; Estep, J.D.; Torre-Amione, G. Full expression of Cardiomyopathy Is Partly Dependent on B-Cells: A Pathway That Involves Cytokine Activation, Immunoglobulin Deposition, and Activation of Apoptosis. J. Am. Heart Assoc. 2016, 5, e002484. [Google Scholar] [CrossRef]
- Lund, F.E. Cytokine-producing B lymphocytes—Key regulators of immunity. Curr. Opin. Immunol. 2008, 20, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guérin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laroumanie, F.; Douin-Echinard, V.; Pozzo, J.; Lairez, O.; Tortosa, F.; Vinel, C.; Delage, C.; Calise, D.; Dutaur, M.; Parini, A.; et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 2014, 129, 2111–2124. [Google Scholar] [CrossRef] [PubMed]
- Nevers, T.; Salvador, A.M.; Grodecki-Pena, A.; Knapp, A.; Velázquez, F.; Aronovitz, M.; Kapur, N.K.; Karas, R.H.; Blanton, R.M.; Alcaide, P. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circ. Heart Fail. 2015, 8, 776–787. [Google Scholar] [CrossRef]
- Tae, Y.H.; Youn, J.; Lee, J.; Park, S.; Chi, H.S.; Lee, J.; Choi, C.; Park, S.; Choi, D.; Ha, J.W.; et al. Characterization of CD8(+)CD57(+) T cells in patients with acute myocardial infarction. Cell Mol. Immunol. 2015, 12, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Ogawa, M.; Suzuki, J.; Hirata, Y.; Nagai, R.; Isobe, M. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int. Heart J. 2011, 52, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Yang, J.; Dong, M.; Zhang, K.; Tu, E.; Gao, Q.; Chen, W.; Zhang, C.; Zhang, Y. Regulatory T cells in cardiovascular diseases. Nat. Rev. Cardiol. 2016, 13, 167–179. [Google Scholar] [CrossRef]
- Hofmann, U.; Beyersdorf, N.; Weirather, J.; Podolskaya, A.; Bauersachs, J.; Ertl, G.; Kerkau, T.; Frantz, S. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 2012, 125, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.L.; Hsiao, Y.W.; Tsai, Y.N.; Lin, S.F.; Liu, S.H.; Lin, Y.J.; Lo, L.W.; Chung, F.P.; Chao, T.F.; Hu, Y.F.; et al. Interleukin-17 enhances cardiac ventricular remodeling via activating MAPK pathway in ischemic heart failure. J. Mol. Cell. Cardiol. 2018, 122, 69–79. [Google Scholar] [CrossRef]
- Pistulli, R.; König, S.; Drobnik, S.; Kretzschmar, D.; Rohm, I.; Lichtenauer, M.; Fritzenwanger, M.; Mall, G.; Mall, G.; Figulla, H.R.; et al. Decrease in dendritic cells in endomyocardial biopsies of human dilated cardiomyopathy. Eur. J. Heart Fail. 2013, 15, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, T.; Honda, S.; Sugano, Y.; Matsuyama, T.A.; Ohta-Ogo, K.; Asaumi, Y.; Ikeda, Y.; Kusano, K.; Ishihara, M.; Yasuda, S.; et al. Decreased myocardial dendritic cells is associated with impaired reparative fibrosis and development of cardiac rupture after myocardial infarction in humans. J. Am. Heart Assoc. 2014, 3, e000839. [Google Scholar] [CrossRef]
- Kallikourdis, M.; Martini, E.; Carullo, P.; Sardi, C.; Roselli, G.; Greco, C.M.; Vignali, D.; Riva, F.; Ormbostad Berre, A.M.; Stølen, T.O.; et al. T cell costimulation blockade blunts pressure overload-induced heart failure. Nat. Commun. 2017, 8, 14680. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.P.; Erskine, J.; Zhang, W.W.; Zheng, R.H.; Zhang, L.H.; Duron, G.; Gendreau, J.; Zhao, ZQ. Recruitment of macrophages from the spleen contributes to myocardial fibrosis and hypertension induced by angiotensin II. J. Renin Angiotensin Aldosterone Syst. 2017, 18, 1470320317706653. [Google Scholar] [CrossRef]
- Brenes-Castro, D.; Castillo, E.C.; Vázquez-Garza, E.; Torre-Amione, G.; García-Rivas, G. Temporal Frame of Immune Cell Infiltration during Heart Failure Establishment: Lessons from Animal Models. Int. J. Mol. Sci. 2018, 19, 3719. [Google Scholar] [CrossRef]
- Westermann, D.; Lindner, D.; Kasner, M. Cardiac Inflammation Contributes to Changes in the Extracellular Matrix in Patients With Heart Failure and Normal Ejection Fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Glezeva, N.; Voon, V.; Watson, C.; Horgan, S.; McDonald, K.; Ledwidge, M.; Baugh, J. Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: Evidence of M2 macrophage activation in disease pathogenesis. J. Card. Fail. 2015, 21, 167–177. [Google Scholar] [CrossRef]
- Yusuf, S.; Pfeffer, M.A.; Swedberg, K.; Granger, C.B.; Held, P.; McMurray, J.J.; Michelson, E.L.; Olofsson, B.; Ostergren, J.; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: The CHARM-Preserved Trial. Lancet 2003, 362, 777–781. [Google Scholar] [CrossRef]
- Massie, B.M.; Carson, P.E.; McMurray, J.J.; Komajda, M.; McKelvie, R.; Zile, M.R.; Anderson, S.; Donovan, M.; Iverson, E.; Staiger, C.; et al. Irbesartan in patients with heart failure and preserved ejection fraction. N. Engl. J. Med. 2008, 359, 2456–2467. [Google Scholar] [CrossRef]
- Pitt, B.; Pfeffer, M.A.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med. 2014, 370, 1383–1392. [Google Scholar] [CrossRef]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef]
- Heinzel, F.R.; Hohendanner, F.; Jin, G.; Sedej, S.; Edelmann, F. Myocardial hypertrophy and its role in heart failure with preserved ejection fraction. J. Appl. Physiol. 2015, 119, 1233–1242. [Google Scholar] [CrossRef]
- Zile, M.R.; Baicu, C.F.; Ikonomidis, J.S.; Stroud, R.E.; Nietert, P.J.; Bradshaw, A.D.; Slater, R.; Palmer, B.M.; Van Buren, P.; Meyer, M.; et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: Contributions of collagen and titin. Circulation 2015, 131, 1247–1259. [Google Scholar] [CrossRef]
- Takimoto, E. Cyclic GMP-dependent signaling in cardiac myocytes. Circ. J. 2012, 76, 1819–1825. [Google Scholar] [CrossRef]
- Kovács, Á.; Alogna, A.; Post, H.; Hamdani, N. Is enhancing cGMP-PKG signalling a promising therapeutic target for heart failure with preserved ejection fraction? Neth. Heart J. 2016, 24, 268–274. [Google Scholar] [CrossRef] [Green Version]
- LeWinter, M.M.; Granzier, H.L. Cardiac titin and heart disease. J. Cardiovasc. Pharmacol. 2014, 63, 207–212. [Google Scholar] [CrossRef] [PubMed]
- LeWinter, M.M.; Zile, M.R. Could Modification of Titin Contribute to an Answer for Heart Failure With Preserved Ejection Fraction? Circulation 2016, 134, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Kruger, M.; Kotter, S.; Grützner, A.; Lang, P.; Andresen, C.; Redfield, M.M.; Butt, E.; dos Remedios, C.G.; Linke, W.A. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 2009, 104, 87–94. [Google Scholar] [CrossRef]
- Borbély, A.; Falcao-Pires, I.; van Heerebeek, L.; Hamdani, N.; Edes, I.; Gavina, C.; Leite-Moreira, A.F.; Bronzwaer, J.G.; Papp, Z.; van der Velden, J.; et al. Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ. Res. 2009, 104, 780–786. [Google Scholar] [CrossRef]
- Hamdani, N.; Bishu, K.G.; von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2013, 97, 464–471. [Google Scholar] [CrossRef]
- van Heerebeek, L.; Hamdani, N.; Falcão-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.; Bronzwaer, J.G.; van der Velden, J.; Stienen, G.J.; Laarman, G.J.; Somsen, A.; et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschöpe, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef]
- Wilck, N.; Markó, L.; Balogh, A. Nitric oxide-sensitive guanylyl cyclase stimulation improves experimental heart failure with preserved ejection fraction. JCI Insight 2018, 3, e96006. [Google Scholar] [CrossRef]
- Bishu, K.; Hamdani, N.; Mohammed, S.F.; Kruger, M.; Ohtani, T.; Ogut, O.; Brozovich, F.V.; Burnett, J.C. Jr.; Linke, W.A.; Redfield, M.M. Sildenafil and B-type natriuretic peptide acutely phosphorylate titin and improve diastolic distensibility in vivo. Circulation 2011, 124, 2882–2891. [Google Scholar] [CrossRef]
- Fraccarollo, D.; Galuppo, P.; Motschenbacher, S.; Ruetten, H.; Schäfer, A.; Bauersachs, J. Soluble guanylyl cyclase activation improves progressive cardiac remodeling and failure after myocardial infarction. Cardioprotection over ACE inhibition. Basic Res. Cardiol. 2014, 109, 421. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Fang, F.; Yu, C.M. Noncardiac Comorbidities in Heart Failure With Preserved Ejection Fraction—A Commonly Ignored Fact. Circ. J. 2015, 79, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Sorop, A.; Heinonen, I.; van Kranenburg, M. Multiple common comorbidities produce left ventricular diastolic dysfunction associated with coronary microvascular dysfunction, oxidative stress, and myocardial stiffening. Cardiovasc. Res. 2018, 114, 954–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, D.J.; Somaratne, J.B.; Prior, D.L.; Yii, M.; Kenn, J.F.; Newcomb, A.E.; Kelly, D.J.; Black, M.J. Obesity is associated with lower coronary microvascular density. PLoS ONE 2013, 8, e81798. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Kitzman, D.W.; Borlaug, B.A.; van Heerebeek, L.; Zile, M.R.; Kass, D.A.; Paulus, W.J. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation 2016, 134, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Panahi, M.; Papanikolaou, A.; Torabi, A.; Zhang, J.G.; Khan, H.; Vazir, A.; Hasham, M.G.; Cleland, J.G.F.; Rosenthal, N.A.; Harding, S.E.; et al. Immunmodulatory interventions in myocardial infarction and heart failure: A systematic review of clinical trials and meta-analysis of IL-1 inhibition. Cardiovasc. Res. 2018, 114, 1445–1461. [Google Scholar] [CrossRef]
- Liu, C.; Liu, K. Cardiac outcome prevention effectiveness of glucocorticoids in acute decompensated heart failure. J. Cardiovasc. Pharmacol. 2014, 63, 333–338. [Google Scholar] [CrossRef]
- Mentzelopoulos, S.D.; Malachias, S.; Chamos, C.; Konstantopoulos, D.; Ntaidou, T.; Papastylianou, A.; Kolliantzaki, I.; Theodoridi, M.; Ischaki, H.; Makris, D.; et al. Vasopressin, steroids, and epinephrine and neurologically favorable survival after in-hospital cardiac arrest. JAMA 2013, 310, 270–279. [Google Scholar] [CrossRef]
- Tsai, M.S.; Huang, C.H.; Chang, W.T.; Chen, W.J.; Hsu, C.Y.; Hsieh, C.C.; Yang, C.W.; Chiang, W.C.; Ma, M.H.; Chen, S.C. The effect of hydrocortisone on the outcome of out-of-hospital cardiac arrest patients: A pilot study. Am. J. Emerg. Med. 2007, 25, 318–325. [Google Scholar] [CrossRef]
- Moreira, D.M.; Lueneberg, M.E.; da Silva, R.L.; Fattah, T.; Gottschall, C.A.M. MethotrexaTE THerapy in ST-Segment Elevation MYocardial Infarction: A randomized double-blind, placebo-controlled trial (TETHYS Trial). J. Cardiovasc. Pharmacol. Ther. 2017, 22, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Pradhan, A.D.; Solomon, D.H.; Paynter, N.; Macfadyen, J.; Zaharris, E.; Gupta, M.; Clearfield, M.; Libby, P.; Hasan, A.A.; et al. Rationale and design of the Cardiovascular Inflammation Reduction Trial: A test of the inflammatory hypothesis of atherothrombosis. Am. Heart J. 2013, 166, 199. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.M.; Vieira, J.L.; Gottschall, C.A.M. The effects of methotrexate therapy on the physical capacity of patients with ischemic heart failure: A randomized double-blind, placebo-controlled trial (METIS Trial). J. Card Fail. 2009, 15, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Yingzhong, C.; Lin, C.; Chunbin, W. Clinical effects of cyclosporine A on reperfusion injury in myocardial infarction: A meta-analysis of randomized controlled trials. Springerplus 2016, 5, 1117. [Google Scholar] [CrossRef] [PubMed]
- Gullestad, L.; Orn, S.; Dickstein, K.; Eek, C.; Edvardsen, T.; Aakhus, S.; Askevold, E.T.; Michelsen, A.; Bendz, B.; Skårdal, R.; et al. Intravenous immunoglobulin does not reduce left ventricular remodeling in patients with myocardial dysfunction during hospitalization after acute myocardial infarction. Int. J. Cardiol. 2013, 168, 212–218. [Google Scholar] [CrossRef]
- Gullestad, L.; Aass, H.; Fjeld, J.G.; Wikeby, L.; Andreassen, A.K.; Ihlen, H.; Simonsen, S.; Kjekshus, J.; Nitter-Hauge, S.; Ueland, T.; et al. Immunomodulating therapy with intravenous immunoglobulin in patients with chronic heart failure. Circulation 2001, 103, 220–225. [Google Scholar] [CrossRef] [PubMed]
- McNamara, D.M.; Holubkov, R.; Starling, R.C. Intervention in Myocarditis and Acute Cardiomyopathy (IMAC) Investigators, et al. Controlled trial of intravenous immune globulin in recent-onset dilated cardiomyopathy. Circulation 2001, 103, 2254–2259. [Google Scholar] [CrossRef]
- van Tassell, B.W.; Trankle, C.R.; Canada, J.M.; Carbone, S.; Buckley, L.; Kadariya, D.; Del Buono, M.G.; Billingsley, H.; Wohlford, G.; Viscusi, M.; et al. IL-1 Blockade in Patients With Heart Failure With Preserved Ejection Fraction- Results From DHART2. Circ. Heart Fail. 2018, 11, e005036. [Google Scholar] [CrossRef]
- van Tassell, B.W.; Abouzaki, N.A.; Oddi Erdle, C.; Carbone, S.; Trankle, C.R.; Melchior, R.D.; Turlington, J.S.; Thurber, C.J.; Christopher, S.; Dixon, D.L.; et al. Interleukin-1 blockade in acute decompensated heart failure. J. Cardiovasc. Pharmacol. 2016, 67, 544–551. [Google Scholar] [CrossRef]
- Morton, A.C.; Rothman, A.M.; Greenwood, J.P.; Gunn, J.; Chase, A.; Clarke, B.; Hall, A.S.; Fox, K.; Foley, C.; Banya, W.; et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA Heart Study. Eur. Heart J. 2015, 36, 377–384. [Google Scholar] [CrossRef]
- Abbate, A.; Kontos, M.C.; Grizzard, J.D.; Biondi-Zoccai, G.G.; van Tassell, B.W.; Robati, R.; Roach, L.M.; Arena, R.A.; Roberts, C.S.; Varma, A.; et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot Study). Am. J. Cardiol. 2010, 105, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy With Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Trankle, C.R.; Canada, J.M.; Cei, L.; Abouzaki, N.; Oddi-Erdle, C.; Kadariya, D.; Christopher, S.; Viscusi, M.; Del Buono, M.; Kontos, M.C.; et al. Usefulness of Canakinumab to Improve Exercise Capacity in Patients With Long-Term Systolic Heart Failure and Elevated C-Reactive Protein. Am. J. Cardiol. 2018, 122, 1366–1370. [Google Scholar] [CrossRef]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: A double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef]
- Mann, D.L.; McMurray, J.J.V.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Dijan, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted anticytokine therapy in patients with chronic heart failure: Results of the randomized etanercept worldwide evaluation (RENEWAL). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T.; Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-α, in patients with moderate-to-severe heart failure. Circulation 2003, 107, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Fattouch, K.; Bianco, G.; Speziale, G.; Sampognaro, R.; Lavalle, C.; Guccione, F.; Dioguardi, P.; Ruvolo, G. Beneficial effects of C1 esterase inhibitor in ST-elevation myocardial infarction in patients who underwent surgical reperfusion: A randomised double-blind study. Eur. J. Cardiothorac. Surg. 2007, 32, 326–332. [Google Scholar] [CrossRef]
- Testa, L.; Van Gaal, W.J.; Bhindi, R.; Biondi-Zoccai, G.G.; Abbate, A.; Agostini, P.; Porto, I.; Andreotti, F.; Crea, F.; Banning, A.P. Pexelizumab in ischemic heart disease: A systematic review and meta-analysis on 15,196 patients. J. Thorac. Cardiovasc. Surg. 2008, 136, 884–893. [Google Scholar] [CrossRef] [Green Version]
- Pasupathy, S.; Tavella, R.; Grover, S.; Raman, B.; Procter, N.E.K.; Du, Y.T.; Mahadavan, G.; Stafford, I.; Heresztyn, T.; Holmes, A.; et al. Early use of N-Acetylcysteine (NAC) with nitrate therapy in patients undergoing primary percutaneous coronary intervention for ST-segment elevation myocardial infarction reduces myocardial infarct size (the NACIAM trial). Circulation 2017, 136, 894–903. [Google Scholar] [CrossRef]
- Pieske, B.; Maggioni, A.P.; Lam, C.S.P.; Pieske-Kraigher, E.; Filippatos, G.; Butler, J.; Ponikowski, P.; Shah, S.J.; Solomon, S.D.; Scalise, A.V.; et al. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: Results of the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED EF (SOCRATES-PRESERVED) study. Eur. Heart J. 2017, 38, 1119–1127. [Google Scholar] [CrossRef]
- Gao, Q.; Yang, B.; Guo, Y.; Zheng, F. Efficacy of adenosine in patients with acute myocardial infarction undergoing primary percutaneous coronary intervention: A PRISMA-compliant meta-analysis. Medicine 2015, 94, e1279. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D.R.; Savage, M.; LaBlanche, J.M.; Grip, L.; Serruys, P.W.; Fitzgerald, P.; Fischman, D.; Goldberg, S.; Brinker, J.A.; Zeiher, A.M.; et al. Results of Prevention of REStenosis with Tranilast and its Outcomes (PRESTO) trial. Circulation 2002, 106, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ogai, A.; Nakatani, S.; Hashimura, K.; Kanzaki, H.; Komamura, K.; Asakura, M.; Asanuma, H.; Kitamura, S.; Tomoike, H.; et al. Impact of blockade of histamine H2 receptors on chronic heart failure revealed by retrospective and prospective randomized studies. J. Am. Coll. Cardiol. 2006, 48, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Faxon, D.P.; Gibbons, R.J.; Chronos, N.A.; Gurbel, P.A.; Sheehan, F.; Halt MI Investigators. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: The results of the HALT-MI study. J. Am. Coll. Cardiol. 2002, 40, 1199–1204. [Google Scholar] [CrossRef]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Genestier, L.; Paillot, R.; Fournel, S.; Ferraro, C.; Miossec, P.; Revillard, J.P. Immunosuppressive properties of methotrexate: apoptosis and clonal deletion of activated peripheral T cells. J. Clin. Invest. 1998, 102, 322–328. [Google Scholar] [CrossRef]
- Tedesco, D.; Haragsim, L. Cyclosporine: A review. J. Transpl. 2012, 2012, 230386. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Boston-Griffiths, E.A.; Yellon, D.M. Cyclosporin A and cardioprotection: From investigative tool to therapeutic agent. Br. J. Pharmacol. 2012, 165, 1235–1245. [Google Scholar] [CrossRef]
- Gelfand, E.W. Intravenous Immune Globulin in Autoimmune and Inflammatory Diseases. N. Engl. J. Med. 2012, 367, 2015–2025. [Google Scholar] [CrossRef]
- Ridker, P.M.; Howard, C.P.; Walter, V.; Everett, B.; Libby, P.; Hensen, J.; Thuren, T.; CANTOS Pilot Investigative Group. Effects of interleukin-1 inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation 2012, 126, 2739–2748. [Google Scholar] [CrossRef]
- Harouki, N.; Nicol, L.; Remy-Jouet, I.; Henry, J.P.; Dumesnil, A.; Lejeune, A.; Renet, S.; Golding, F.; Djerada, Z.; Wecker, D.; et al. The IL-1β Antibody Gevokizumab Limits Cardiac Remodeling and Coronary Dysfunction in Rats With Heart Failure. JACC Basic Transl. Sci. 2017, 2, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Torre-Amione, G.; Warren, M.S.; Whitmore, J.; Soran, O.Z.; Feldman, A.M.; Mann, D.L. Results of targeted anti-tumor necrosis factor therapy with etanercept (ENBREL) in patients with advanced heart failure. Circulation 2001, 103, 1044–1047. [Google Scholar] [CrossRef] [PubMed]
- Deswal, A.; Bozkurt, B.; Seta, Y.; Parilti-Eiswirth, S.; Hayes, F.A.; Blosch, C.; Mann, D.L. Safety and efficacy of a soluble P75 tumor necrosis factor receptor (Enbrel, etanercept) in patients with advanced heart failure. Circulation 1999, 99, 3224–3226. [Google Scholar] [CrossRef] [PubMed]
- Fraccarollo, D.; Galuppo, P.; Sieweke, J.T.; Napp, L.C.; Grobbecker, P.; Bauersachs, J. Efficacy of mineralcorticoid receptor antagonism in the acute myocardial infarction phase: Eplerenone versus spironolactone. ESC Heart Fail. 2015, 2, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Sliwa, K.; Skudicky, D.; Candy, G.; Badenhorst, D.; Libhaber, C.; Norton, G.; Skudicky, D.; Sareli, P. Effects of pentoxifylline on cytokine profiles and left ventricular performance in patients with decompensated congestive heart failure secondary to idiopathic dilated cardiomyopathy. Am. J. Cardiol. 2002, 90, 1118–1122. [Google Scholar] [CrossRef]
- Shao, T.; Zhang, Y.; Tang, R.; Zhang, H.; Wang, Q.; Yang, Y.; Liu, T. Effects of milrinone on serum IL-6, TNF-α, Cys-C and cardiac functions of patients with chronic heart failure. Exp. Ther. Med. 2018, 16, 4162–4166. [Google Scholar] [CrossRef]
- Stanciu, A.E.; Vatasescu, R.G.; Stanciu, M.M.; Iorgulescu, C.; Vasile, A.I.; Dorobantu, M. Cardiac resynchronization therapy in patients with chronic heart failure is associated with anti-inflammatory and anti-remodeling effects. Clin. Biochem. 2013, 46, 230–234. [Google Scholar] [CrossRef]
- Yu, L.; Huang, B.; Po, S.S.; Tan, T.; Wang, M.; Zhou, L.; Meng, G.; Yuan, S.; Zhou, X.; Li, X.; et al. Low-Level Tragus Stimulation for the Treatment of Ischemia and Reperfusion Injury in Patients With ST-Segment Elevation Myocardial Infarction: A Proof-of-Concept Study. JACC Cardiovasc. Interv. 2017, 10, 1511–1520. [Google Scholar] [CrossRef]
- Tompkins, B.A.; Balkan, W.; Winkler, J.; Gyöngyösi, M.; Goliasch, G.; Fernández-Avilés, F.; Hare, J.M. Preclinical Studies of Stem Cell Therapy for Heart Disease. Circ. Res. 2018, 122, 1006–1020. [Google Scholar] [CrossRef]
- Huyer, L.D.; Montgomery, M.; Zhao, Y.; Xiao, Y.; Conant, G.; Korolj, A.; Radisic, M. Biomaterial based cardiac tissue engineering and its applications. Biomed. Mater. 2015, 10, 034004. [Google Scholar] [CrossRef] [PubMed]
- Hulot, J.S.; Ishikawa, K.; Hajjar, R.J. Gene therapy for the treatment of heart failure: Promise postponed. Eur. Heart J. 2016, 37, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- de Souza Rebouças, J.; Santos-Magalhães, N.S.; Formiga, F.R. Cardiac Regeneration using Growth Factors: Advances and Challenges. Arq. Bras. Cardiol. 2016, 107, 271–275. [Google Scholar]
- Rapezzi, C.; Lorenzini, M.; Longhi, S.; Milandri, A.; Gagliardi, C.; Bartolomei, I.; Salvi, F.; Maurer, M.S. Cardiac amyloidosis: The great pretender. Heart Fail. Rev. 2015, 20, 117–124. [Google Scholar] [CrossRef]
- Maurer, M.S.; Elliott, P.; Comenzo, R.; Semigran, M.; Rapezzi, C. Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation 2017, 135, 1357–1377. [Google Scholar] [CrossRef] [PubMed]
- Dungu, J.N.; Papadopoulou, S.A.; Wykes, K.; Mahmood, I.; Marshall, J.; Valencia, O.; Fontana, M.; Whelan, C.J.; Gilmore, J.D.; Hawkins, P.N.; et al. Afro-Caribbean heart failure in the United Kingdom: Cause, outcomes, and ATTR V122I cardiac amyloidosis. Circ. Heart Fail. 2016, 9, e003352. [Google Scholar] [CrossRef] [PubMed]
- Quarta, C.C.; Buxbaum, J.N.; Shah, A.M.; Falk, R.H.; Claggett, B.; Kitzman, D.W.; Mosley, T.H.; Butler, K.R.; Boerwinkle, E.; Solomon, S.D. The amyloidogenic V122I transthyretin variant in elderly black Americans. N. Engl. J. Med. 2015, 372, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Horibata, Y.; Shono, M.; Misumi, Y.; Oshima, T.; Su, Y.; Tasaki, M.; Shinriki, S.; Kawahara, S.; Jono, H.; et al. Clinicopathological features of senile systemic amyloidosis: An ante- and post-mortem study. Mod. Pathol. 2011, 24, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Benson, M.D.; Dyck, P.J.; Grogan, M.; Coelho, T.; Cruz, M.; Berk, J.L.; Planté-Bordeneuve, V.; Schmidt, H.H.J.; Merlini, G. Diagnosis, prognosis and therapy of transthyretin amyloidosis. J. Am. Coll. Cardiol. 2015, 66, 2451–2466. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, G.; Berni, R. Plasma retinol-binding protein: Structure and interactions with retinol retinoids, and transthyretin. Vitam. Horm. 2004, 69, 271–295. [Google Scholar]
- Quintas, A.; Saraiva, M.J.; Brito, R.M. The tetrameric protein transthyretin dissociates to a non-native monomer in solution. A novel model for amyloidogenesis. J. Biol. Chem. 1999, 274, 32943–32949. [Google Scholar] [CrossRef] [PubMed]
- Planté-Bordeneuve, V.; Carayol, J.; Ferreira, A.; Adams, D.; Clerget-Darpoux, F.; Misrahi, M.; Said, G.; Bonaïti-Pellié, C. Genetic study of transthyretin amyloid neuropathies: Carrier risks among French and Portuguese families. J. Med. Genet. 2003, 40, e120. [Google Scholar] [CrossRef] [PubMed]
- Hellman, U.; Alarcon, F.; Lundgren, H.E.; Suhr, O.B.; Bonaïti-Pellié, C.; Planté-Bordeneuve, V. Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid 2008, 15, 181–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, M.D. Hereditary amyloidosis. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D., Beaudet, A.L., Vogelstein, B., Kinzler, K.W., Antonarakis, S.E., Ballabio, A.K., Gibson, M., Mitchell, G., Eds.; McGraw-Hill Medical: New York, NY, USA, 2007; Available online: https://ommbid.mhmedical.com/content.aspx?sectionid=62657674&bookid=971&Resultclick=2 (accessed on 22 February 2019).
- Sekijima, Y.; Yazaki, M.; Ueda, M.; Koike, H.; Yamada, M.; Ando, Y. First nationwide survey on systemic wild-type ATTR amyloidosis in Japan. Amyloid 2018, 25, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; Adams, D.; Kristen, A.; Grogan, M.; González-Duarte, A.; Maurer, M.S.; Merlini, G.; Damy, T.; Slama, M.S.; Brannagan, T.H., 3rd; et al. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis: An analysis of the APOLLO study. Circulation 2018, 139, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Valshnaw, A.K.; Maraganore, J.M.; Vest, J.; Brodsky, J. ALNY—2017 RNAi Roundtable: Revusiran investigation Results: Edited Transcript. Available online: http://www.alnylam.com/wp-content/uploads/2017/08/ Revusiran_Roundtable_Transcript_2017-08-09-1.pdf (accessed on 24 February 2019).
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Benson, M.D.; Ackermann, E.J.; Monia, B.P. Treatment of transthyretin cardiomyopathy with a TTR-specific antisense oligonucleotide (IONIS-TTRRx). Amyloid 2017, 24, 134–135. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliot, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA 2013, 310, 2658–2667. [Google Scholar] [CrossRef]
- Castaño, A.; Helmke, S.; Alvarez, J.; Delisle, S.; Maurer, M.S. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail. 2012, 18, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Karlstedt, E.; Jimenez-Zepeda, V.; Howlett, J.G.; White, J.A.; Fine, N.M. Clinical Experience With the Use of Doxycycline and Ursodeoxycholic Acid for the Treatment of Transthyretin Cardiac Amyloidosis. J. Card Fail. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wixner, J.; Pilebro, B.; Lundgren, H.E.; Olsson, M.; Anan, I. Effect of doxycycline and ursodeoxycholic acid on transthyretin amyloidosis. Amyloid 2017, 24, 78–79. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y. Hereditary Transthyretin Amyloidosis; Last Updated 20 December 2018; GeneReviews® NCBI Bookshelf; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 1993–2019; Available online: https://www.ncbi.nlm.nih.gov/books/NBK1194/?report=printable (accessed on 22 February 2019).
- Nagakawa, M.; Sekijima, Y.; Yazaki, M.; Tojo, K.; Yoshinaga, T.; Doden, T.; Koyama, J.; Yanagisawa, S.; Ikeda, S. Carpal tunnel syndrome: A common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid 2016, 23, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Maurer, M.S.; Suhr, O.B. THAOS: The Transthyretin Amyloidosis Outcomes Survey: Initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr. Med. Res. Opin. 2013, 29, 63–76. [Google Scholar] [CrossRef]
- Connors, L.H.; Sam, F.; Skinner, M.; Salinaro, F.; Sun, F.; Ruberg, F.L.; Berk, J.L.; Seldin, D.C. Heart failure resulting from age-related cardiac amyloid disease associated with wild-type transthyretin: A prospective, observational cohort study. Circulation 2016, 133, 282–290. [Google Scholar] [CrossRef]
- González-López, E.; Gagliardi, C.; Dominguez, F.; Quarta, C.C.; de Haro-Del Moral, F.J.; Milandri, A.; Salas, C.; Cinelli, M.; Cobo-Marcos, M.; Lorenzini, M.; et al. Clinical characteristics of wild-type transthyretin cardiac amyloidosis: Disproving myths. Eur. Heart J. 2017, 38, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, F.; Baldasseroni, S.; Bergesio, F.; Perlini, S.; Salinaro, F.; Padeletti, L.; Attanà, P.; Paoletti Perini, A.; Moggi Pignone, A.; Grifoni, E.; et al. Echocardiographic and biohumoral characteristics in patients with AL and TTR amyloidosis at diagnosis. Clin. Cardiol. 2015, 38, 69–75. [Google Scholar] [CrossRef]
- Granstam, S.O.; Rosengren, S.; Vedin, O.; Kero, T.; Sörensen, J.; Carlson, K.; Flachskampf, F.A.; Wikström, G. Evaluation of patients with cardiac amyloidosis using echocardiography, ECG and right heart catherization. Amyloid 2013, 20, 27–33. [Google Scholar] [CrossRef]
- Rapezzi, C.; Quarta, C.C.; Guidalotti, P.L.; Pettinato, C.; Fanti, S.; Leone, O.; Ferlini, A.; Longhi, S.; Lorenzini, M.; Reggiani, L.B.; et al. Rolle of 99mTc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. J. Am. Coll. Cardiol. Img. 2011, 4, 659–670. [Google Scholar] [CrossRef]
- Longhi, S.; Guidalotti, P.L.; Quarta, C.C.; Gagliardi, C.; Milandri, A.; Lorenzini, M.; Potena, L.; Leone, O.; Bartolomei, I.; Pastorelli, F.; et al. Identification of TTR-related subclinical amyloidosis with 99mTc-DPD scintigraphy. J. Am. Coll. Cardiol. Img. 2014, 7, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Aljaroudi, W.A.; Desai, M.Y.; Tang, W.H.; Phelan, D.; Cerqueira, M.D.; Jaber, W.A. Role of imaging in the diagnosis and management of patients with cardiac amyloidosis: State of the art review and focus on emerging nuclear techniques. J. Nucl. Cardiol. 2014, 21, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Glaudemans, A.W.; van Rheenan, R.W.; van den Berg, M.P.; Noordzij, W.; Koole, M.; Blokzijl, H.; Dierckx, R.A.; Slart, R.H.; Hazenberg, B.P. Bone scintigraphy with 99mtechnetium-hydroxymethylene diphosphonate allows early diagnosis of cardiac involvement in patients with transthyretin-derived systemic amyloidosis. Amyloid 2014, 21, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Park, M.A.; Padera, R.F.; Belanger, A.; Dubey, S.; Hwang, D.H.; Veeranna, V.; Falk, R.H.; Di Carli, M.F.; Dorbala, S. 18F-florbetapir binds specifically to myocardial light chain and transthyretin amyloid deposits: Autoradiography study. Circ. Cardiovasc. Imaging 2015, 8, e002954. [Google Scholar] [CrossRef] [PubMed]
- Perugini, E.; Guidalotti, P.L.; Salvi, F.; Cooke, R.M.; Pettinato, C.; Riva, L.; Leone, O.; Farsad, M.; Ciliberti, P.; Bacchi-Reggiani, L.; et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-disphosphono-1,2-propanodicarboxylic acid scintigraphy. J. Am. Coll. Cardiol. 2005, 46, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Dungu, J.N.; Valencia, O.; Pinney, J.H.; Gibbs, S.D.; Rowczenio, D.; Gilbertson, J.A.; Lachmann, H.J.; Wechalekar, A.; Gillmore, J.D.; Whelan, C.J.; et al. CMR-based differentiation of AL and ATTR cardiac amyloidosis. J. Am. Coll. Cardiol. Img. 2014, 7, 133–142. [Google Scholar] [CrossRef]
- Sado, D.M.; Flett, A.S.; Banypersad, S.M.; White, S.K.; Maestrini, V.; Quarta, G.; Lachmann, R.H.; Murphy, E.; Mehta, A.; Hughes, D.A.; et al. Cardiovascular magnetic resonance measurement of myocardial extracellular volume in health and disease. Heart 2012, 98, 1436–1441. [Google Scholar] [CrossRef]
- Fontana, M.; Pica, S.; Reant, P.; Abdel-Gadir, A.; Treibel, T.A.; Banypersad, S.M.; Maestrini, V.; Barcella, W.; Rosmini, S.; Bulluck, H.; et al. Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2015, 132, 1570–1579. [Google Scholar] [CrossRef]
- Kristen, A.V.; Maurer, M.S.; Rapezzi, C.; Mundayat, R.; Suhr, O.B.; Damy, T.; THAOS Investigators. Impact of genotype and phenotype on cardiac biomarkers in patients with transthyretin amyloidosis—Report from the Transthyretin Amyloidosis Outcome Survey (THAOS). PLoS ONE 2017, 12, e0173086. [Google Scholar] [CrossRef]
- Ikeda, S.; Sekijima, Y.; Tojo, K.; Koyama, J. Diagnostic value of abdominal wall fat pad biopsy in senile systemic amyloidosis. Amyloid 2011, 18, 211–215. [Google Scholar] [CrossRef]
- Jamet, M.P.; Gnemmi, V.; Hachulla, É.; Dhaenens, C.M.; Bouchindhomme, B.; Delattre, C.; Glowacki, F.; Hatron, P.Y.; Lacour, A.; Lamblin, N.; et al. Distinctive Patterns of Transthyretin Amyloid in Salivary Tissue: A Clinicopathologic Study of 92 Patients With Amyloid-containing Minor Salivary Gland Biopsies. Am. J. Surg. Pathol. 2015, 39, 1035–1044. [Google Scholar] [CrossRef]
- Fine, N.M.; Arruda-Olson, A.M.; Dispenzieri, A.; Zeldenrust, S.R.; Gertz, M.A.; Kyle, R.A.; Swiecicki, P.L.; Scott, C.G.; Grogan, M. Yield of noncardiac biopsy for the diagnosis of transthyretin cardiac amyloidosis. Am. J. Cardiol. 2014, 113, 1723–1727. [Google Scholar] [CrossRef]
- de Fernández Larrea, C.; Verga, L.; Morbini, P.; Klersy, C.; Lavatelli, F.; Foli, A.; Obici, L.; Milani, P.; Capello, G.L.; et al. A practical approach to the diagnosis of systemic amyloidoses. Blood 2015, 125, 2239–2244. [Google Scholar] [CrossRef] [Green Version]
- Gilbertson, J.A.; Theis, J.D.; Vrana, J.A.; Lachmann, H.; Wechalekar, A.; Whelan, C.; Hawkins, P.N.; Dogan, A.; Gillmore, J.D. A comparison of immunohistochemistry and mass spectrometry for determining the amyloid fibril protein from formalin-fixed biopsy tissue. J. Clin. Pathol. 2015, 68, 314–317. [Google Scholar] [CrossRef]
- Schmidt, H.H.J.; Barroso, F.; González-Duarte, A.; Conceiҫão, I.; Obici, L.; Keohane, D.; Amass, L. Management of asymptomatic gene carriers of transthyretin familial amyloid polyneuropathy. Muscle Nerve 2016, 54, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Obici, L.; Kuks, J.B.; Buades, J.; Adams, D.; Suhr, O.B.; Coelho, T.; Kyriakides, T.; European Network for TTR-FAP (ATTReuNET). Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr. Opin. Neurol. 2016, 29, 27–35. [Google Scholar] [CrossRef]
- Yamamoto, S.; Wilczek, H.E.; Nowak, G.; Larsson, M.; Oksanen, A.; Iwata, T.; Gjertsen, H.; Söderdahl, G.; Wikström, L.; Ando, Y.; et al. Liver transplantation for familial amyloidotic polyneuropathy (FAP): A single-center experience over 16 years. Am. J. Transpl. 2007, 7, 2597–2604. [Google Scholar] [CrossRef]
- Carvalho, A.; Rocha, A.; Lobato, L. Liver transplantation in transthyretin amyloidosis: Issues and challenges. Liver Transpl. 2015, 21, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Yazaki, M.; Mitsuhashi, S.; Tokuda, T.; Kametani, F.; Takei, Y.I.; Koyama, J.; Kawamorita, A.; Kanno, H.; Ikeda, S.I. Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am. J. Transpl. 2007, 7, 235–242. [Google Scholar] [CrossRef]
- Banerjee, D.; Roeker, L.E.; Grogan, M.; Swiecicki, P.; Poterucha, J.; Heimbach, J.; Zeldenrust, S.; Gertz, M.; Edwards, B.; Daly, R.; et al. Outcomes of Patients With Familial Transthyretin Amyloidosis After Liver Transplantation. Progress Transpl. 2017, 27, 246–250. [Google Scholar] [CrossRef]
- Suhr, O.B.; Coelho, T.; Buades, J.; Pouget, J.; Conceiҫão, I.; Berk, J.; Schmidt, H.; Waddington-Cruz, M.; Campistol, J.M.; Bettencourt, B.R.; et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: A phase II multi-dose study. Orphanet J. Rare Dis. 2015, 10, 109. [Google Scholar] [CrossRef]
- Coelho, T.; Maia, L.F.; Martins da Silva, A.; Waddington-Cruz, M.; Planté-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceiҫão, I.M.; Schmidt, H.H.; et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 2012, 79, 785–792. [Google Scholar] [CrossRef]
- Planté-Bordeneuve, V.; Gorram, F.; Salhi, H.; Nordine, T.; Ayache, S.S.; Le Corvoisier, P.; Azoulay, D.; Feray, C.; Damy, T.; Lefaucheur, J.P. Long-term treatment of transthyretin familial amyloid polyneuropathy with tafamidis: A clinical and neurophysiological study. J. Neurol. 2017, 264, 268–276. [Google Scholar] [CrossRef]
- Penchala, S.C.; Connelly, S.; Wang, Y.; Park, M.S.; Zhao, L.; Baranczak, A.; Rappley, I.; Vogel, H.; Liedtke, M.; Witteles, R.M.; et al. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc. Natl. Acad. Sci. USA 2013, 110, 9992–9997. [Google Scholar] [CrossRef] [Green Version]
- Higaki, J.N.; Chakrabartty, A.; Galant, N.J.; Hadley, K.C.; Hammerson, B.; Nijjar, T.; Torres, R.; Tapia, J.R.; Salmans, J.; Barbour, R.; et al. Novel conformation-specific monoclonal antibodies against amyloidogenic forms of transthyretin. Amyloid 2106, 23, 86–97. [Google Scholar] [CrossRef]
- Richards, D.B.; Cookson, L.M.; Barton, S.V.; Liefaard, L.; Lane, T.; Hutt, D.F.; Ritter, J.M.; Fontana, M.; Moon, J.C.; Gillmore, J.D.; et al. Repeat doses of antibody to serum amyloid P component clear amyloid deposits in patients with systemic amyloidosis. Sci. Transl. Med. 2018, 10, eaan3128. [Google Scholar] [CrossRef] [Green Version]


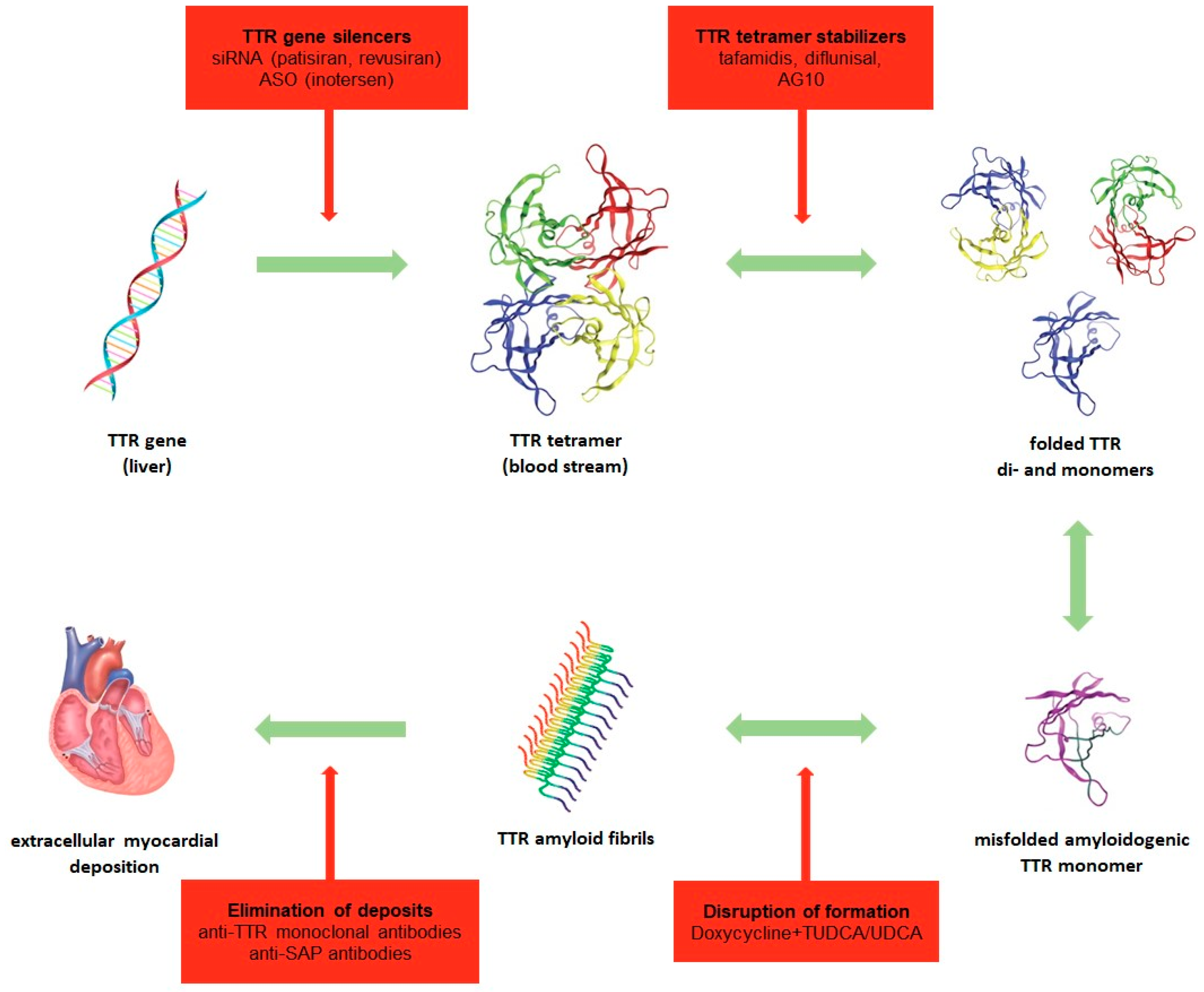{kind=link}
| Immunosuppression | |||||
|---|---|---|---|---|---|
| Trial | Study Population | n | Treatment | Follow-Up | Primary Outcome |
| Corticosteroids | |||||
| COPE-ADHF [88] | Acute decompensated HF | 102 | Dexamethasone or prednisolone 20 mg IV once daily; 1 mg/kg daily (max. 60 mg) for 7 days then tapered off vs. standard treatment | 19 months | Reduced cardiac mortality, improvement of dyspnea and global clinical status |
| Mentzelopoulos et al. [89] | Cardiac arrest | 268 | Methylprednisolone 40 mg IV once and hydrocortisone 300 mg IV daily for 7 days then tapered off vs. saline placebo | 2 months | Improved rate of ROSC, survival to discharge and neurological outcome |
| Tsai et al. [90] | Cardiac arrest | 97 | Hydrocortisone 100 mg IV during resuscitation vs. saline placebo | 7 days | Higher ROSC rate, no difference in survival and hospitality discharge rates |
| Methotrexate | |||||
| TETHYS [91] | STEMI | 84 | 0.05 mg/kg + 0.05 mg/kg/h for 6 days vs. placebo | 3 months | No difference in mortality, coronary blood flow, infarct size, cardiac markers or reinfarction, worsened LVEF at 3 months |
| CIRT [92] | Prior MI and either type 2 diabetes or metabolic syndrome | 7000 | Target dose 15–20 mg/week | 3–5 years | Results pending |
| METIS [93] | Ischemic congestive HF | 50 | 7.5 mg/week for 12 weeks | 3 months | Tendency toward improved NYHA, no difference in 6MWT or MACE |
| Cyclosporine A | |||||
| Yingzhong et al. [94] (meta-analysis): CYCLE (2016) Cung et al. (2015) Ghaffari et al. (2013) Mewton et al. (2010) Piot et al. (2008) | Acute MI / STEMI | ∑ 1250 | 2.5 mg/kg IV vs. placebo | 6 months, Cung 12 months | No difference in all-cause mortality or adverse clinical events, no significant improvement of LVEF or infarct size |
| IVIg | |||||
| Gullestad et al. [95] | Acute MI treated by PCI | 62 | 0.4 g/kg once daily for 5 days then 0.4 g/kg monthly for 26 weeks vs. placebo | 6 months | No effect on LV remodeling or function and inflammatory markers after completed maintenance therapy |
| Gullestad et al. [96] | Congestive HF and LVEF <40% | 40 | 0.4 g/kg once daily for 5 days then 0.4 g/kg monthly for 5 months vs. placebo | 6 months | Marked rise of anti-inflammatory markers and significant increase in LVEF |
| IMAC [97] | Recent onset of idiopathic DCM and LVEF <40% | 62 | 1 g/kg IVIG for 2 days vs. placebo | 12 months | No difference in LVEF improvement |
| Immunomodulation | |||||
| IL-1 inhibitors | |||||
| DHART 2 [98] | HFpEF and CRP >2 mg/L | 31 | Anakinra 100 mg sc daily vs. placebo for 12 weeks vs. placebo | 24 weeks | No improvement in cardiorespiratory fitness |
| Van Tassell et al. [99] | Acute decompensated HF, LVEF <40% and CRP ≥5 mg/L | 30 | Anakinra 100 mg sc twice daily for 3 days followed by once daily for 11 days vs. placebo | 14 days | Reduction in systemic inflammatory response, no evaluation of cardiac function/clinical outcomes |
| MRC-ILA Heart Study [100] | NSTEMI <48 h from onset of chest pain | 182 | Anakinra 100 mg sc for 14 days vs. placebo | 12 months | Reduction in inflammatory markers, higher rate of MACE at 1 year |
| VCU-ART [101] | Acute MI | 10 | Anakinra 100 mg sc daily for 14 days | 14 weeks | Favorably affected LV end-systolic and -diastolic volume index |
| Everett et al. [102] Ridker et al. [103] (CANTOS) | Prior MI and high-sensitivity CRP ≥2 mg/L | 10,061 | Canakinumab 50, 150, or 300 mg sc once every 3 months vs. placebo | 3.7 years | Dose-dependent reduction in hospitalization for HF and the composite of hospitalization or HF-related mortality, lower rate of recurrent cardiovascular events |
| Trankle et al. [104] (CANTOS sub study) | Prior MI, high-sensitivity CRP ≥2 mg/L and LVEF <50% | 30 | Canakinumab 50, 150 or 300 mg sc once every 3 months vs. placebo | 12 months | Improvement of cardiorespiratory fitness and LVEF |
| IL-6 receptor antagonist | |||||
| Kleveland et al. [105] | NSTEMI | 117 | Tocilizumab 280 mg IV single dose vs. placebo prior to coronary angiography | 6 months | Attenuation of inflammatory response (hs-CRP, leukocytes, hs-TNT) |
| TNF-α inhibitors | |||||
| RENEWAL [106] (RECOVER and RENAISSANCE) | Chronic HF, NYHA II-IV and LVEF ≤30% | 925 + 1123 | Etanercept 25 mg sc once or twice a week vs. placebo;etanercept 25 mg sc twice or three times per week vs. placebo | 24 weeks | No effect on clinical status, hospitalization due to chronic HF or mortality |
| ATTACH [107] | Chronic HF, NYHA III-IV and LVEF ≤35% | 150 | Infliximab 5 or 10 mg/kg or placebo at 0, 2 and 6 weeks | 28 weeks | No improvement after short-term treatment, higher risk of hospitalization due to HF and death under 10 mg/kg |
| Complement inhibitors | |||||
| Fattouch et al. [108] (C1 esterase inhibitor) | STEMI undergoing emergent CABG | 80 | C1-INH 1000 UI vs. placebo | 48 h | Improved cardiac function (CI, SV) and haemodynamics without impact on early mortality rate |
| Testa et al. [109] (meta-analysis of C5-inhibitor, 6 studies) | STEMI or elective CABG | ∑ 15,915 | Pexelizumab 2 mg/kg + 0.05 mg/kg/h for 24 days; pexelizumab 2 mg/kg or 2 mg/kg + 0.05 mg/kg/h for 20 days | 7 days, 3 months, 6 months | In STEMI no benefit in MACE, MI, stroke or heart failure; in CABG 26% reduction in risk of death |
| Targeting ROS and NO-cGMP-PKG signaling | |||||
| NACIAM [110] | STEMI | 112 | High-dose N-acetylcysteine (29 g over 2 days) with background low-dose nitroglycerin (7.2 mg over 2 days) vs. placebo | 3 months | Increased myocardial salvage and reduced infarct size, clinical outcomes not assessed |
| SOCRATES-PRESERVED [111] | HFpEF (LVEF ≥45%) | 477 | Vericiguat once daily at 1.25 or 2.5 mg fixed doses, or 5 or 10 mg titrated from a 2.5 mg starting dose, or placebo for 12 weeks | 12 weeks | No change in NT-proBNP or left atrial volume, improvement in quality of life |
| Targeting adaptive immunity | |||||
| Gao et al. [112] (meta-analysis) | Acute MI | 1736 | Adenosine in varying doses | No improvement of LVEF, all-cause mortality, cardiovascular mortality or re-infarction after PCI | |
| PRESTO [113] (mast cell stabilizer) | PCI of at least one vessel stenosis | 11,484 | Tranilast 300 mg or 450 mg twice daily oral for 1 month or 3 months | 9 months | No improvement of mortality, MACE or target vessel revascularization |
| Kim et al. [114] (histamine H2 receptor antagonist) | Symptomatic congestive HF | 50 | Famotidine 30 mg daily for 6 months vs. teprenone | 6 months | Improved both cardiac symptoms, ventricular remodeling (LVEDV/LVESV) and MACE |
| HALT-MI [115] (CD11/CD18 integrin inhibitor) | STEMI within 6 h of onset of chest pain | 420 | Hu23F2G (Leukoarrest) 0.3 or 1 mg/kg IV bolus or placebo | 1 month | No difference in infarct size, mortality or MACE |
| wtATTR | hATTR | |
|---|---|---|
| Prevalence | Unknown prevalence, higher than thus far assumed, probably very frequent and perhaps leading form of amyloidosis | <1:100,000 |
| Pathogenesis | Sporadic misfolding | Point mutations, most frequent: Val50Met (~73%) and Val142Ile (~4%) |
| Age | >60 years, especially in elderly >80 years, rarely diagnosed during life | At younger age <60 years (30–50 years), depending on mutation |
| Sex | Male predominance | Male predominance with more aggressive phenotype |
| Clinical course | Often asymptomatic | Dependent on mutation and penetrance homozygosity linked to higher incidence, earlier onset and more severe clinical presentation strong genotype-phenotype correlation |
| Affected organs | Dispersed deposition in several organs: primarily cardiac deposition and secondarily neural deposition; eye, kidney and tendon involvement also possible | Val50Met: polyneuropathy, in 43% also cardiac involvement Val142Ile: cardiomyopathy, in 30% also polyneuropathy other mutations with leptomeningeal, ophthalmological and nephrological involvement |
| Cardiac injury |
| |
| Extracardiac injury |
|
|
| Diagnostic methods for cardiomyopathy | ECG, echocardiography, cardiac MRI, cardiac scintigraphy | |
| Substance Group | Agent | Trial and Design | Investigated Population | Efficacy Endpoints Regarding Cardiac Status | Pending Approvals/Trials in Planning |
|---|---|---|---|---|---|
| TTR gene silencer | Patisiran | APOLLO Phase III [146,147] Randomized, double-blind, placebo-controlled | 225 patients with FAP; 56% with cardiac involvement (subgroup analysis) Randomized 2:1 0.3 mg/kg patisiran IV or placebo every 3 weeks for 2 years Follow-up 18 months |
| Regulatory approval granted from FAD and EC for the therapy of FAP Vutrisiran vs. patisiran in hATTR (HELIOS-A; currently recruiting) |
| Revusiran | ENDEAVOUR Phase III [148] Randomized, double-blind, placebo-controlled | 206 patients with FAC Revusiran 500 mg SC for 5 days, then weekly for 18 months years vs. placebo | Discontinued due to increase in mortality in the revusiran arm | ||
| Inotersen | NEURO-TTR Phase III [149] Randomized, double-blind, placebo-controlled | 172 patients with FAP stage I and II Randomized 2:1 300 mg SC every 12 h for 1 week, then weekly for 64 weeks vs. placebo Follow-up 15 months |
| Marketing authorization approved from EC for treatment of stage 1+2 PNP in hATTR; regulatory approval from the FDA for FAP | |
| Phase II study [150] open-label, non-randomized | 20 patients with ATTR cardiomyopathy Inotersen 300 mg SC every 12 h for 1 week |
| |||
| CARDIO-TTR | Trial in patients with FAC | Postponed due to increased thrombocytopenia and bleeding in NEURO-TTR | |||
| TTR stabilizer | Tafamidis | ATTR-ACT Phase III [151] Randomized, double-blind, placebo-controlled | 441 patients with Randomization 2:1:2 tafamidis 80 or 20 mg or placebo orally every 24 h for 30 months |
| Extension phase up to 60 months vs. placebo Approval from EC for use in FAP stage I 01/2019 FDA accepts regulatory submissions for review to treat TTR cardiomyopathy |
| Diflunisal | Phase III study [152] | 130 patients with FAP 50% with cardiac involvement at baseline Randomization 1:1 Diflunisal 250 mg orally every 12 h vs. placebo for 24 months |
| Off-label use in FAP Further efficacy trials required | |
| Phase II study [153] Single-arm, open-label | 13 patients with ATTR cardiomyopathy |
| |||
| AG-10 | Phase II study Randomized, double-blind, placebo-controlled (NCT03458130) | 45 patients with ATTR cardiomyopathy (at least 30% hATTR) Randomization 1:1:1 two different doses of AG10 every 12 h or placebo |
| Further efficacy trials expected (e.g., NCT03536767) | |
| Elimination of deposits | Doxycycline + TUDCA/UDCA | Phase II study [154] | 53 patients with ATTR cardiomyopathy treated with doxycycline and ursodiol, follow-up 22 months |
| Further efficacy trials pending |
| Phase II study [155] | 55 patients in patients with ATTR cardiomyopathy Doxycycline 100 mg orally every 12 h for 4 weeks with a pause of 2 weeks, then UDCA 750 mg (500 + 250 mg) orally per day continuously for 12 months |
| |||
| Anti-SAP | Phase II study Open-label, non-randomized, three groups (NCT03044353) | 40 patients Cohort 1 with ATTR cardiomyopathy Cohort 2 AL at >6 months post chemotherapy Cohort 3 newly diagnosed AL Anti-SAP+CPHPC monthly for 6 months; follow up max. 18 months |
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michels da Silva, D.; Langer, H.; Graf, T. Inflammatory and Molecular Pathways in Heart Failure—Ischemia, HFpEF and Transthyretin Cardiac Amyloidosis. Int. J. Mol. Sci. 2019, 20, 2322. https://doi.org/10.3390/ijms20092322
Michels da Silva D, Langer H, Graf T. Inflammatory and Molecular Pathways in Heart Failure—Ischemia, HFpEF and Transthyretin Cardiac Amyloidosis. International Journal of Molecular Sciences. 2019; 20(9):2322. https://doi.org/10.3390/ijms20092322
Chicago/Turabian StyleMichels da Silva, Diana, Harald Langer, and Tobias Graf. 2019. "Inflammatory and Molecular Pathways in Heart Failure—Ischemia, HFpEF and Transthyretin Cardiac Amyloidosis" International Journal of Molecular Sciences 20, no. 9: 2322. https://doi.org/10.3390/ijms20092322
APA StyleMichels da Silva, D., Langer, H., & Graf, T. (2019). Inflammatory and Molecular Pathways in Heart Failure—Ischemia, HFpEF and Transthyretin Cardiac Amyloidosis. International Journal of Molecular Sciences, 20(9), 2322. https://doi.org/10.3390/ijms20092322