Examining Cardiomyocyte Dysfunction Using Acute Chemical Induction of an Ageing Phenotype

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
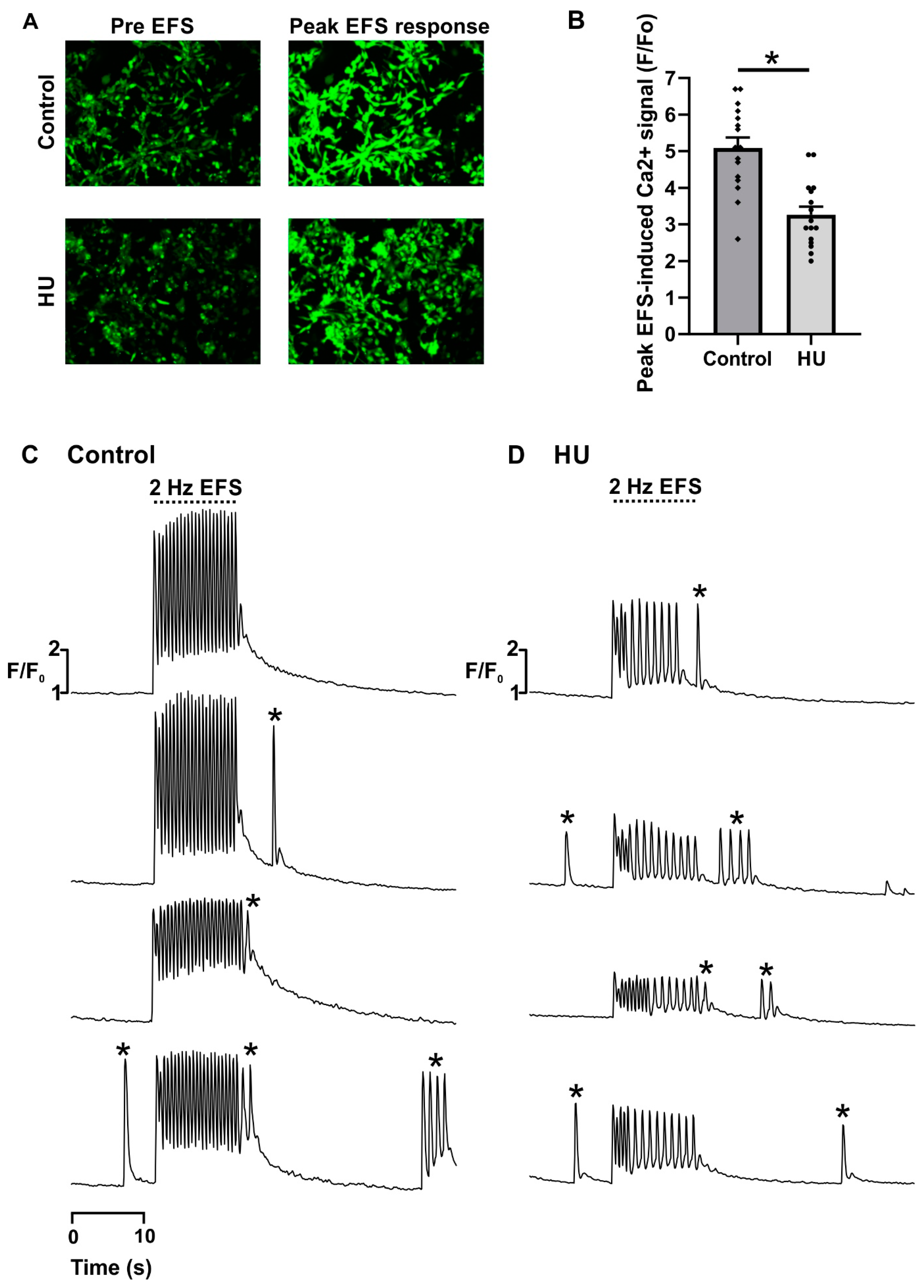
2.1. Altered Ca2+ Signalling in HU-Treated NRVMs
- Ten-second recording of spontaneous activity (‘pre EFS’).
- Ten-second recording during 2 Hz EFS (‘EFS’).
- Ten-second recording of spontaneous activity (‘post EFS’).
- Regular responses. Ca2+ transients that were elicited by every EFS pulse, and had similar pulse-to-pulse amplitudes.
- Alternans. Cells responded to every EFS pulse, but the amplitude of the Ca2+ transients alternated between large and small.
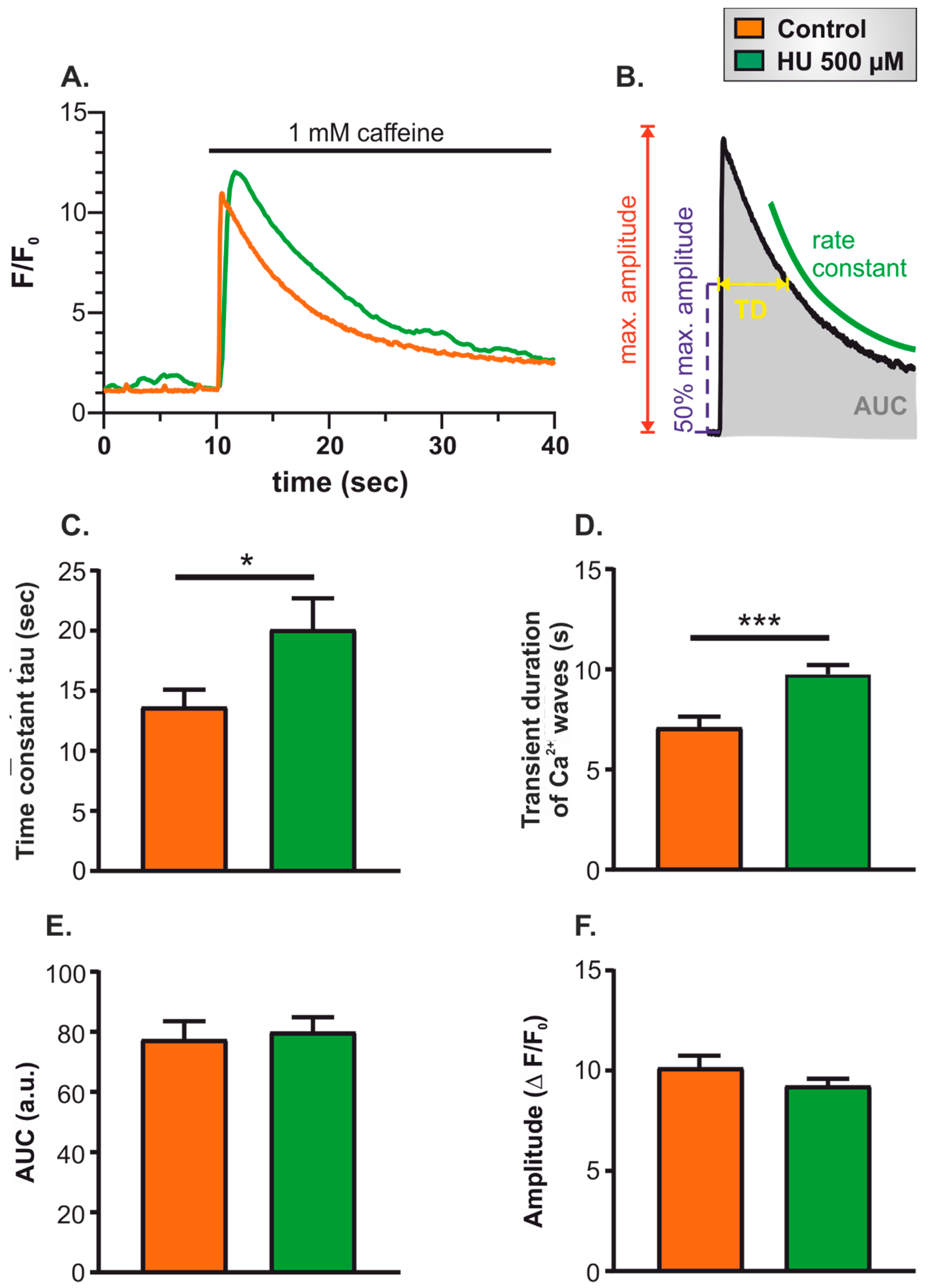
- Twenty-second recording of spontaneous Ca2+ signals in resting cells to ensure that cells were healthy and showing typical levels of spontaneous activity.
- Thirty-second superfusion with Ca2+-free imaging buffer supplemented with the Ca2+ chelator EGTA (500 µM).
- Thirty-second superfusion with 1 mM caffeine in Ca2+-free imaging buffer supplemented with EGTA.
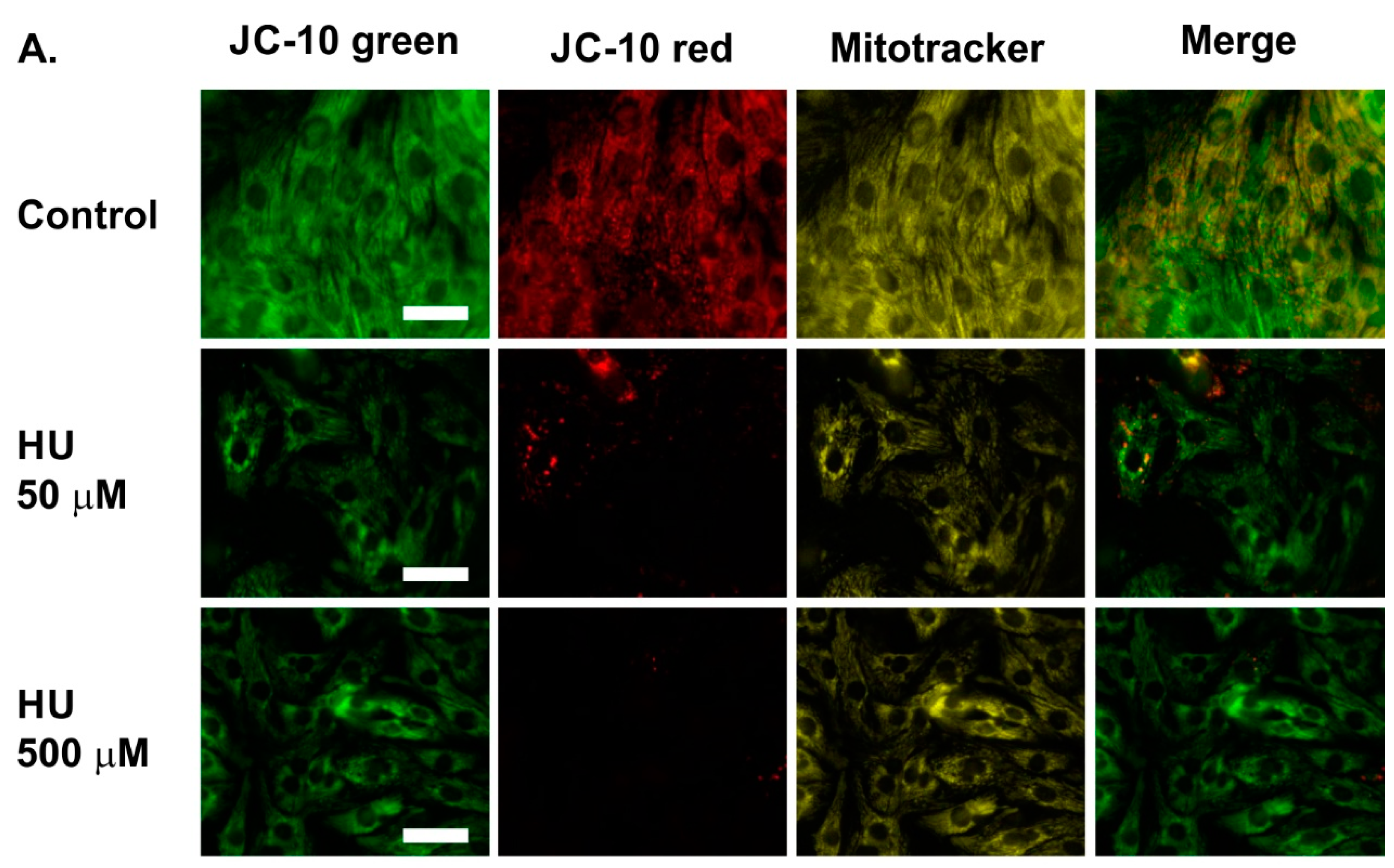
2.2. Altered Mitochondrial Metabolism in HU-Treated NRVMs
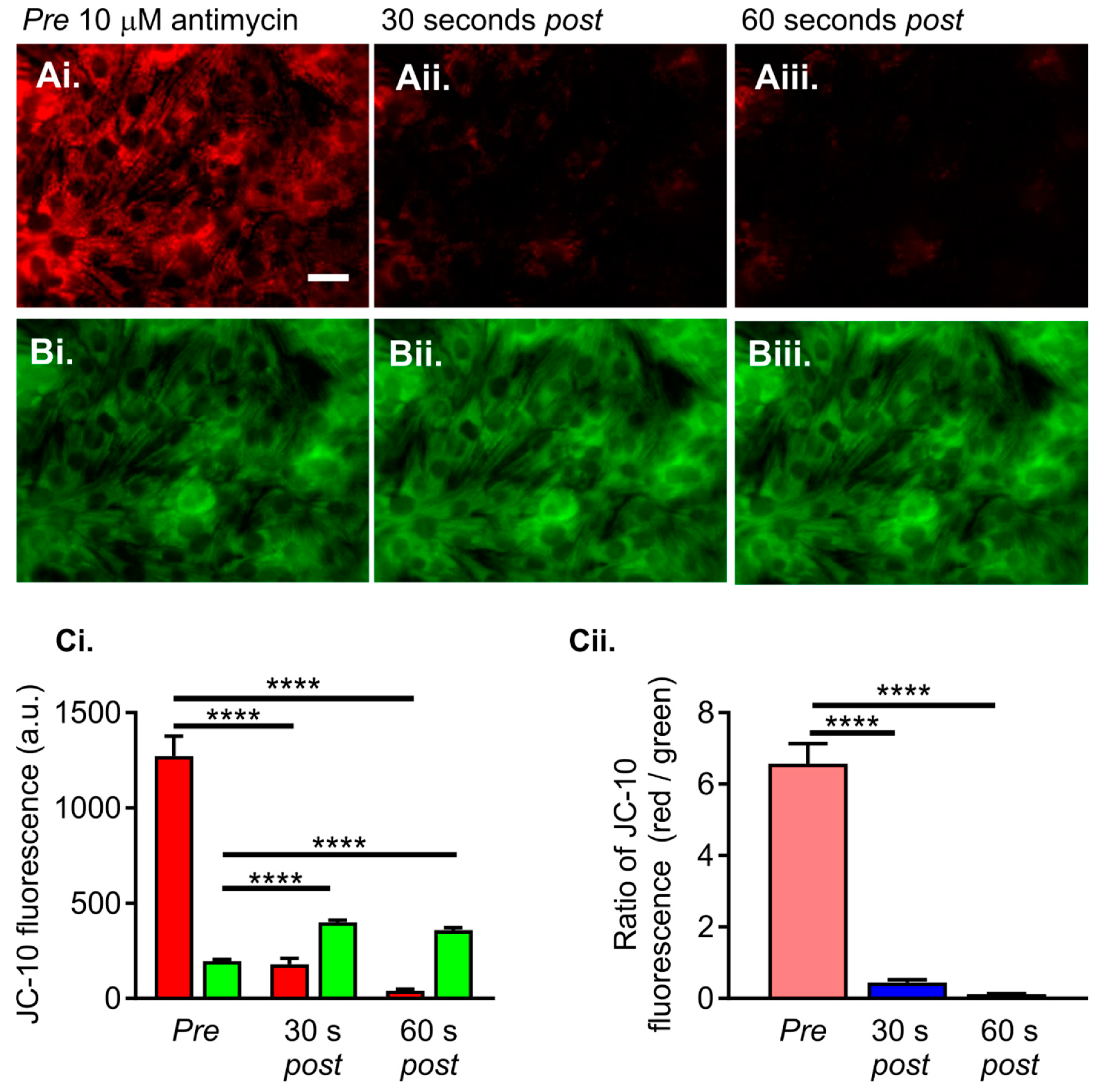
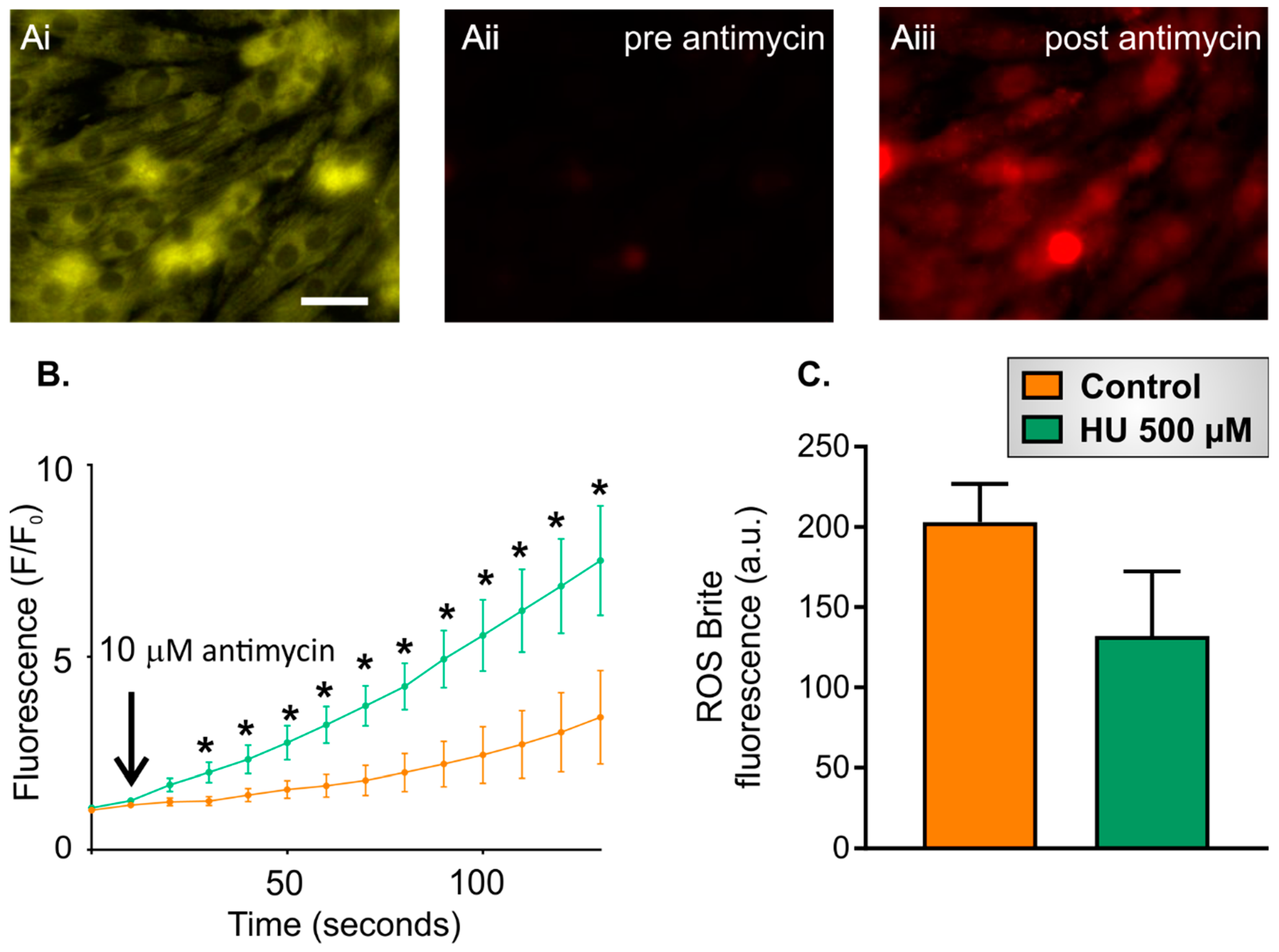
2.3. Altered Reactive Oxygen Species (ROS) Production in HU-Treated NRVMs
- Ten-second recording of ROS Brite fluorescence in control or HU-treated NRVMs.
- Addition of 10 µM antimycin, followed by further imaging of ROS Brite fluorescence for 120 s.
- Basal ROS level. This was calculated as the mean ROS Brite fluorescence in the first five images captured during an imaging experiment.
- Inducible ROS production. This was calculated following the addition of antimycin to the cells by measuring the mean ROS Brite fluorescence in 10-s intervals over the 120-s experimental period. An example of ROS Brite cellular fluorescence in cells treated with antimycin is shown in Figure 7Aiii.
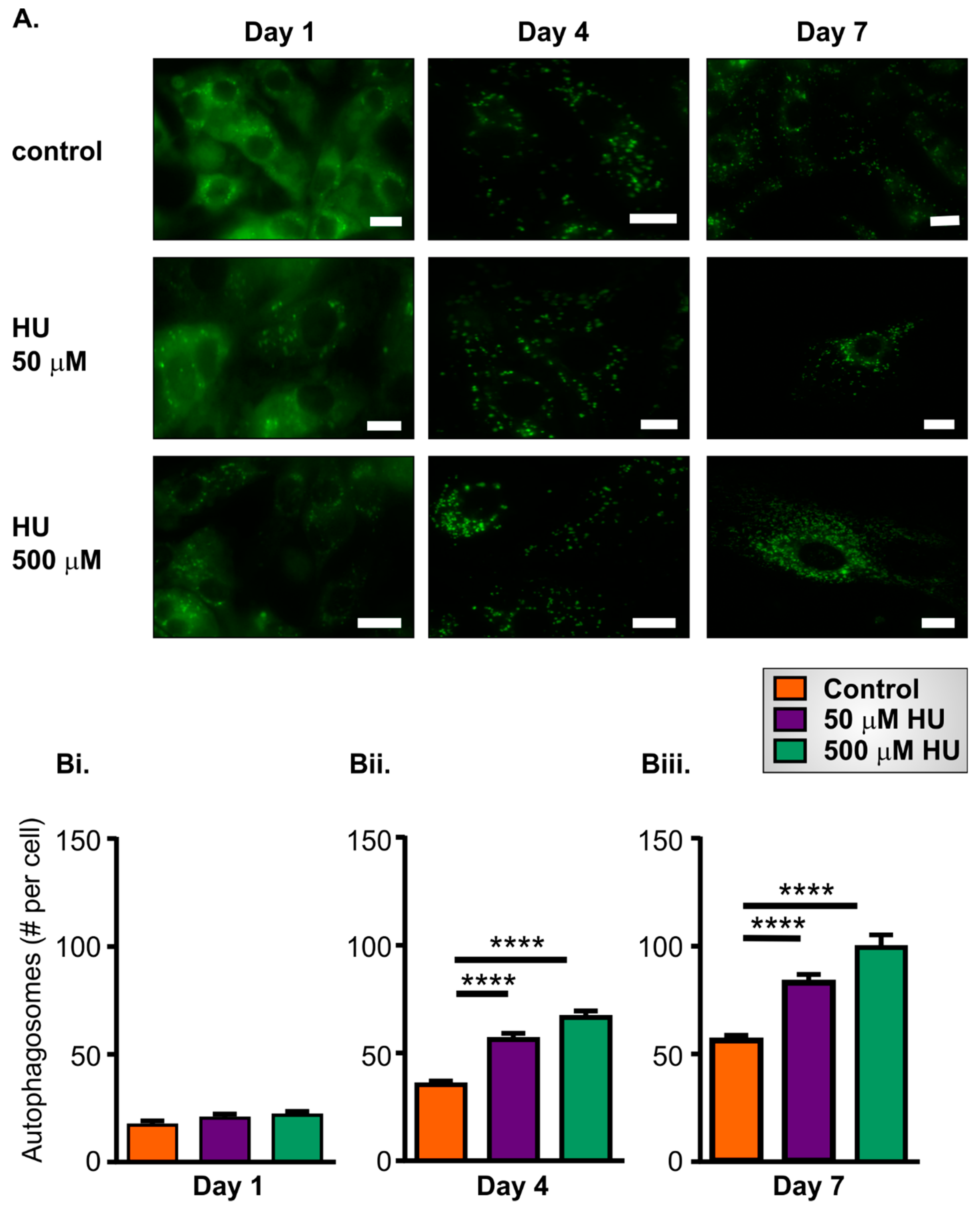
2.4. Increased Autophagy in HU-Treated NRVMs
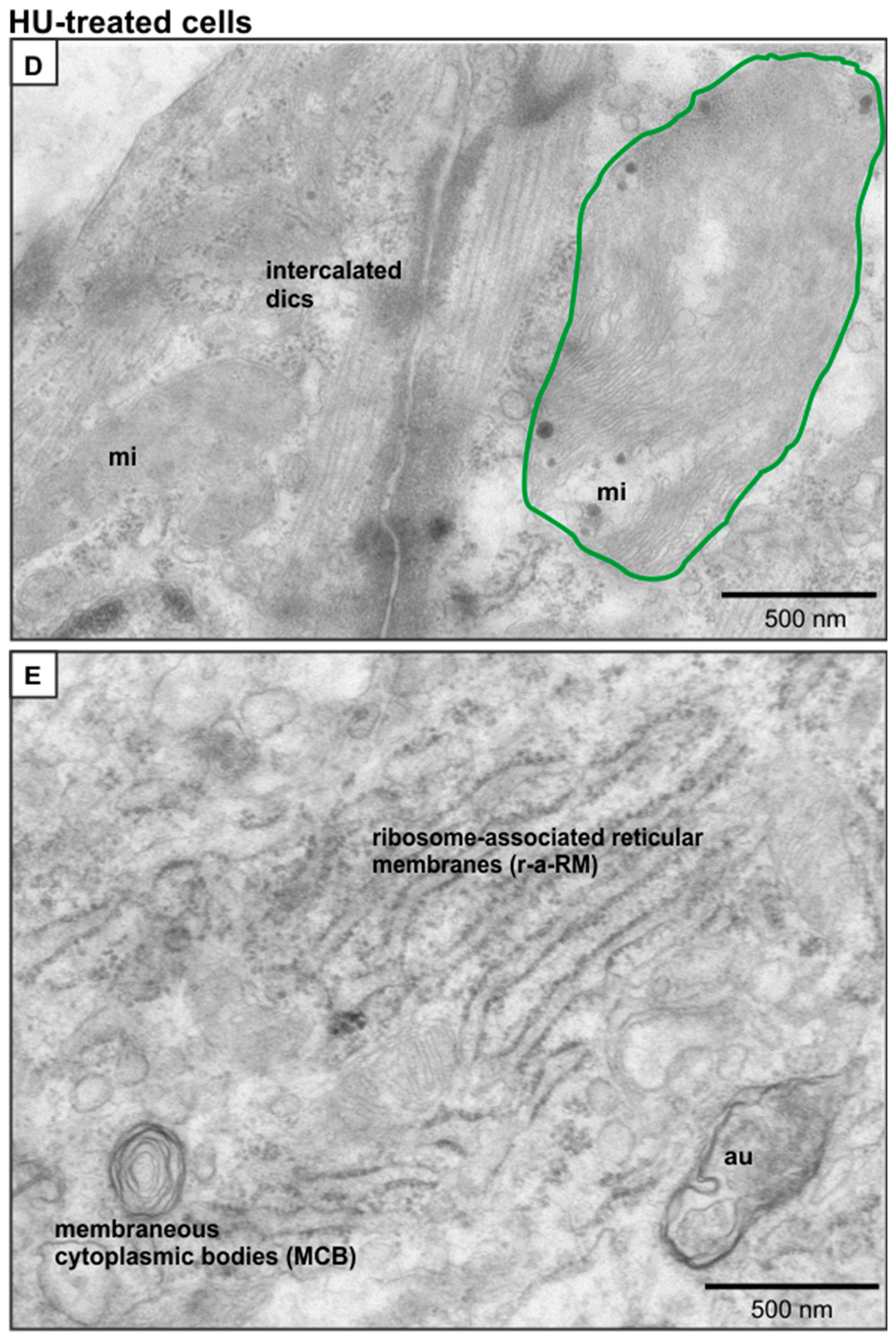
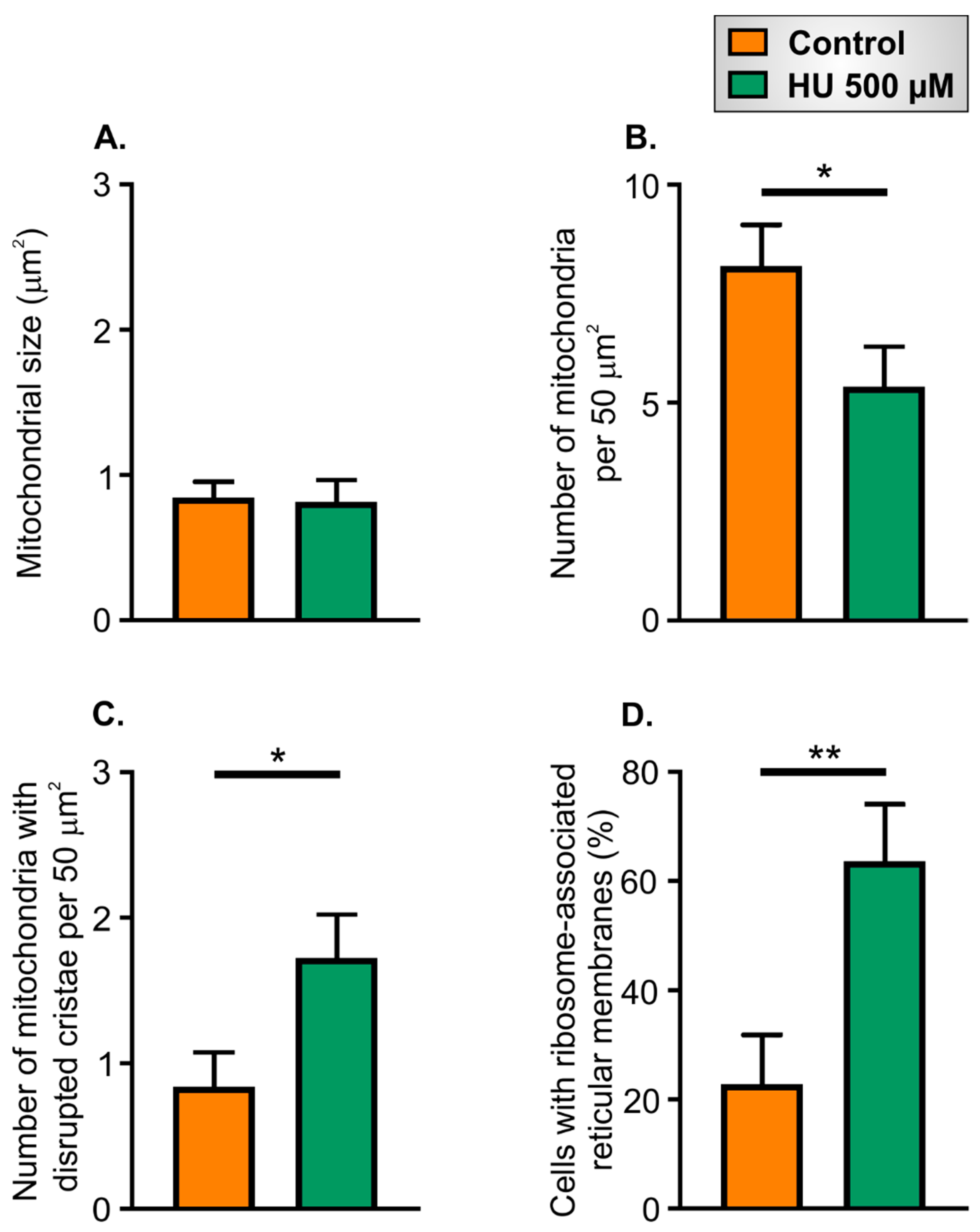
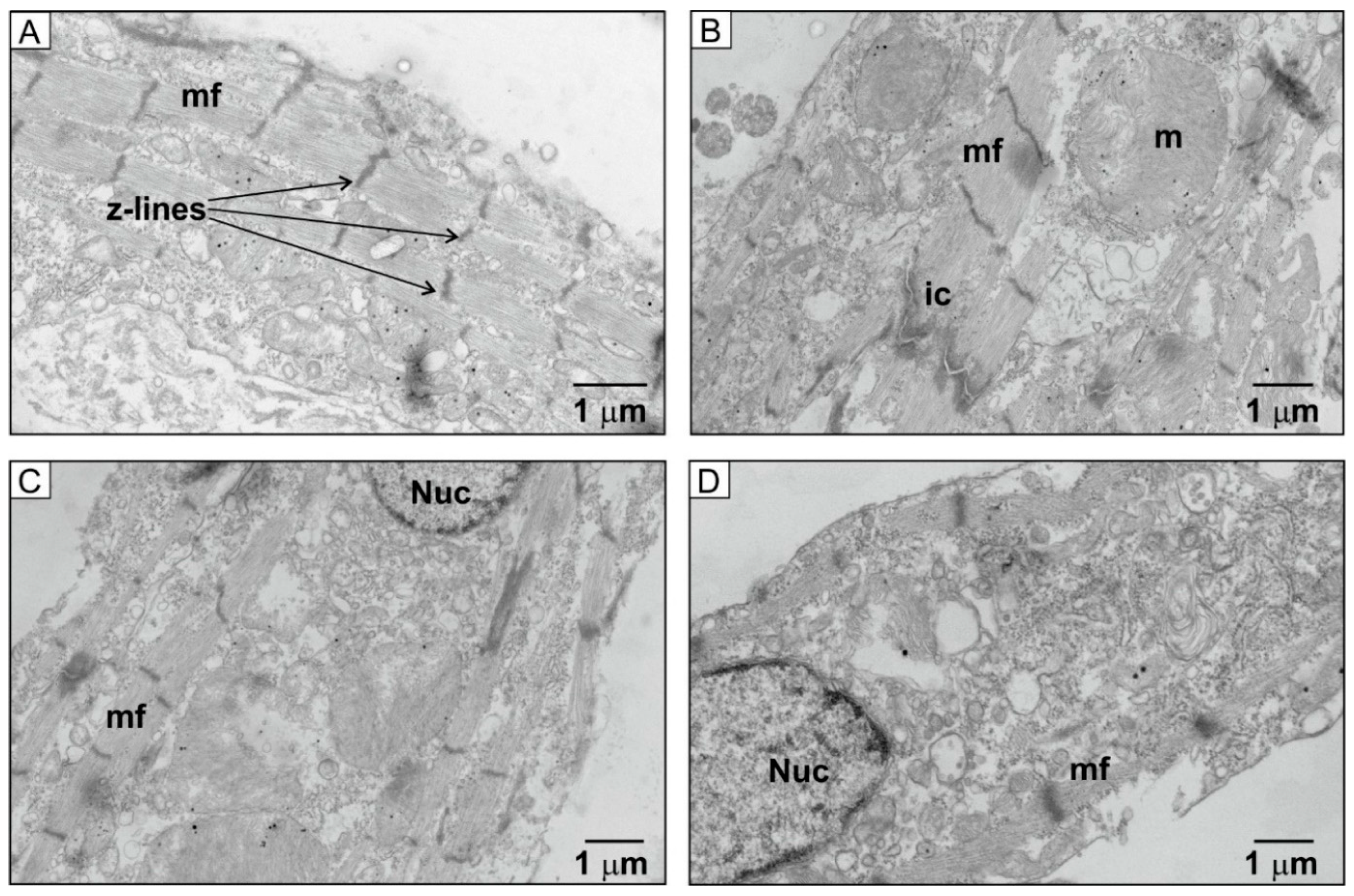
2.5. Ultrastructural Changes in HU-Treated NRVMs
3. Discussion
4. Materials and Methods
4.1. Preparation of NRVMs
4.2. Ca2+ Imaging
4.3. Measurement of the Mitochondrial Membrane Potential
4.4. Measurement of ROS Levels
4.5. Autophagy Assay
4.6. Transmission Electron Microscopy
4.7. Experimental Repeats and Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Mertens, J.; Reid, D.; Lau, S.; Kim, Y.; Gage, F.H. Aging in a Dish: IPSC-Derived and Directly Induced Neurons for Studying Brain Aging and Age-Related Neurodegenerative Diseases. Annu. Rev. Genet. 2018, 52, 271–293. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.; Terentyev, D. Altered Intracellular Calcium Homeostasis and Arrhythmogenesis in the Aged Heart. Int. J. Mol. Sci. 2019, 20, 2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo-Rodriguez, M.; Hernando-Perez, E.; Nunez, L.; Villalobos, C. Amyloid beta Oligomers Increase ER-Mitochondria Ca2+Cross Talk in Young Hippocampal Neurons and Exacerbate Aging-Induced Intracellular Ca2+Remodeling. Front. Cell. Neurosci. 2019, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Calvo, M.; Sanz-Blasco, S.; Caballero, E.; Villalobos, C.; Nunez, L. Susceptibility to excitotoxicity in aged hippocampal cultures and neuroprotection by non-steroidal anti-inflammatory drugs: Role of mitochondrial calcium. J. Neurochem. 2015, 132, 403–417. [Google Scholar] [CrossRef]
- D’Avanzo, C.; Aronson, J.; Kim, Y.H.; Choi, S.H.; Tanzi, R.E.; Kim, D.Y. Alzheimer’s in 3D culture: Challenges and perspectives. BioEssays News Rev. Mol. Cell. Dev. Biol. 2015, 37, 1139–1148. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.M.; Wang, X.L.; Wang, G.M.; Zhang, W.J.; Zhu, L.; Gao, S.; Yang, D.J.; Qin, Y.; Liang, Q.J.; Chen, Y.L.; et al. A stress-induced cellular aging model with postnatal neural stem cells. Cell Death Dis. 2014, 5, e1116. [Google Scholar] [CrossRef]
- Park, J.-I.; Jeong, J.-S.; Han, J.-Y.; Kim, D.-I.; Gao, Y.-H.; Park, S.-C.; Rodgers, G.P.; Kim, I.-H. Hydroxyurea induces a senescence-like change of K562 human erythroleukemia cell. J. Cancer Res. Clin. Oncol. 2000, 126, 455–460. [Google Scholar] [CrossRef]
- Yeo, E.J.; Hwang, Y.C.; Kang, C.M.; Kim, I.H.; Kim, D.I.; Parka, J.S.; Choy, H.E.; Park, W.Y.; Park, S.C. Senescence-like changes induced by hydroxyurea in human diploid fibroblasts. Exp. Gerontol. 2000, 35, 553–571. [Google Scholar] [CrossRef]
- Narath, R.; Ambros, I.M.; Kowalska, A.; Bozsaky, E.; Boukamp, P.; Ambros, P.F. Induction of senescence in MYCN amplified neuroblastoma cell lines by hydroxyurea. Genes Chromosomes Cancer 2007, 46, 130–142. [Google Scholar] [CrossRef]
- Banh, S.; Hales, B.F. Hydroxyurea exposure triggers tissue-specific activation of p38 mitogen-activated protein kinase signaling and the DNA damage response in organogenesis-stage mouse embryos. Toxicol. Sci. Off. J. Soc. Toxicol. 2013, 133, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Krakoff, I.H.; Brown, N.C.; Reichard, P. Inhibition of ribonucleoside diphosphate reductase by hydroxyurea. Cancer Res. 1968, 28, 1559–1565. [Google Scholar] [PubMed]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, G.; Demydenko, K.; Dries, E.; Puertas, R.D.; Jin, X.; Sipido, K.; Roderick, H.L. Calcium Signaling in Cardiomyocyte Function. Cold Spring Harb. Perspect. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.-P.; Su, M.-J.; Lin, J.-L.; Lin, F.-Y.; Tsai, C.-H.; Chen, Y.-S.; Huang, S.K.S.; Tseng, Y.-Z.; Lien, W.-P. Down-regulation of L-type calcium channel and sarcoplasmic reticular Ca2+-ATPase mRNA in human atrial fibrillation without significant change in the mRNA of ryanodine receptor, calsequestrin and phospholamban. J. Am. Coll. Cardiol. 1999, 33, 1231–1237. [Google Scholar] [CrossRef] [Green Version]
- Klein, G. Increased open probability of single cardiac L-type calcium channels in patients with chronic atrial fibrillation Role of phosphatase 2A. Cardiovasc. Res. 2003, 59, 37–45. [Google Scholar] [CrossRef]
- Allessie, M.; Ausma, J.; Schotten, U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc. Res. 2002, 54, 230–246. [Google Scholar] [CrossRef]
- Van der Velden, H.M.; van Kempen, M.J.; Wijffels, M.C.; van Zijverden, M.; Groenewegen, W.A.; Allessie, M.A.; Jongsma, H.J. Altered pattern of connexin40 distribution in persistent atrial fibrillation in the goat. J. Cardiovasc. Electrophysiol. 1998, 9, 596–607. [Google Scholar] [CrossRef]
- Sun, Q.; Tang, M.; Pu, J.; Zhang, S. Pulmonary venous structural remodelling in a canine model of chronic atrial dilation due to mitral regurgitation. Can. J. Cardiol. 2008, 24, 305–308. [Google Scholar] [CrossRef] [Green Version]
- Boldt, A.; Scholl, A.; Garbade, J.; Resetar, M.E.; Mohr, F.W.; Gummert, J.F.; Dhein, S. ACE-inhibitor treatment attenuates atrial structural remodeling in patients with lone chronic atrial fibrillation. Basic Res. Cardiol. 2006, 101, 261–267. [Google Scholar] [CrossRef]
- Denham, N.C.; Pearman, C.M.; Caldwell, J.L.; Madders, G.W.P.; Eisner, D.A.; Trafford, A.W.; Dibb, K.M. Calcium in the Pathophysiology of Atrial Fibrillation and Heart Failure. Front. Physiol. 2018, 9, 1380. [Google Scholar] [CrossRef] [Green Version]
- Rietdorf, K.; Masoud, S.; McDonald, F.; Sanderson, M.J.; Bootman, M.D. Pulmonary vein sleeve cell excitation-contraction-coupling becomes dysynchronized by spontaneous calcium transients. Biochem. Soc. Trans. 2015, 43, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Remodelling Ca2+ signalling systems and cardiac hypertrophy. Biochem. Soc. Trans. 2006, 34, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Gasparova, I.; Kubatka, P.; Opatrilova, R.; Caprnda, M.; Filipova, S.; Rodrigo, L.; Malan, L.; Mozos, I.; Rabajdova, M.; Nosal, V.; et al. Perspectives and challenges of antioxidant therapy for atrial fibrillation. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ausma, J.; Litjens, N.; Lenders, M.H.; Duimel, H.; Mast, F.; Wouters, L.; Ramaekers, F.; Allessie, M.; Borgers, M. Time course of atrial fibrillation-induced cellular structural remodeling in atria of the goat. J. Mol. Cell. Cardiol. 2001, 33, 2083–2094. [Google Scholar] [CrossRef] [Green Version]
- Ausma, J.; Wijffels, M.; Thoné, F.; Wouters, L.; Allessie, M.; Borgers, M. Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation 1997, 96, 3157–3163. [Google Scholar] [CrossRef]
- Thijssen, V.L.; Ausma, J.; Liu, G.S.; Allessie, M.A.; van Eys, G.J.; Borgers, M. Structural changes of atrial myocardium during chronic atrial fibrillation. Cardiovasc. Pathol. 2000, 9, 17–28. [Google Scholar] [CrossRef]
- Schotten, U. The L-type Ca2+-channel subunits α1C and β2 are not downregulated in atrial myocardium of patients with chronic atrial fibrillation. J. Mol. Cell. Cardiol. 2003, 35, 437–443. [Google Scholar] [CrossRef]
- Gaborit, N.; Steenman, M.; Lamirault, G.; Le Meur, N.; Le Bouter, S.; Lande, G.; Leger, J.; Charpentier, F.; Christ, T.; Dobrev, D.; et al. Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation 2005, 112, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Trafford, A.W.; Sibbring, G.C.; Diaz, M.E.; Eisner, D.A. The effects of low concentrations of caffeine on spontaneous Ca release in isolated rat ventricular myocytes. Cell Calcium 2000, 28, 269–276. [Google Scholar] [CrossRef]
- Díaz, M.E.; Trafford, A.W.; O’Neill, S.C.; Eisner, D.A. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J. Physiol. 1997, 501, 3. [Google Scholar] [CrossRef]
- Greensmith, D.J.; Galli, G.L.J.; Trafford, A.W.; Eisner, D.A. Direct measurements of SR free Ca reveal the mechanism underlying the transient effects of RyR potentiation under physiological conditions. Cardiovasc. Res. 2014, 103, 554–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Peng, Y.; Zheng, J.; Zheng, B.; Jin, X.; Liu, H.; Wang, Y.; Tang, X.; Huang, T.; Jiang, P.; et al. A deafness-associated tRNAAsp mutation alters the m1G37 modification, aminoacylation and stability of tRNAAsp and mitochondrial function. Nucleic Acids Res. 2016, 44, 10974–10985. [Google Scholar] [CrossRef] [Green Version]
- Kustiawan, P.M.; Lirdprapamongkol, K.; Palaga, T.; Puthong, S.; Phuwapraisirisan, P.; Svasti, J.; Chanchao, C. Molecular mechanism of cardol, isolated from Trigona incisa stingless bee propolis, induced apoptosis in the SW620 human colorectal cancer cell line. BMC Pharmacol. Toxicol. 2017, 18, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Gordan, R.; Wen, H.; Fefelova, N.; Zang, W.J.; Xie, L.H. Modulation of intracellular calcium waves and triggered activities by mitochondrial ca flux in mouse cardiomyocytes. PLoS ONE 2013, 8, e80574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauvezin, C.; Ayala, C.; Braden, C.R.; Kim, J.; Neufeld, T.P. Assays to monitor autophagy in Drosophila. Methods 2014, 68, 134–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Cuervo, A.M.; Seglen, P.O. Methods for Monitoring Autophagy from Yeast to Human. Autophagy 2007, 3, 181–206. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, C.R.; Gahl, W.A. Lysosomal storage diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef] [Green Version]
- Terman, A.; Dalen, H.; Eaton, J.W.; Neuzil, J.; Brunk, U.T. Aging of Cardiac Myocytes in Culture: Oxidative Stress, Lipofuscin Accumulation, and Mitochondrial Turnover. Ann. N. Y. Acad. Sci. 2004, 1019, 70–77. [Google Scholar] [CrossRef]
- Bootman, M.D.; Smyrnias, I.; Thul, R.; Coombes, S.; Roderick, H.L. Atrial cardiomyocyte calcium signalling. Biochim. Biophys. Acta 2011, 1813, 922–934. [Google Scholar] [CrossRef] [Green Version]
- Geissler, S.; Textor, M.; Kuhnisch, J.; Konnig, D.; Klein, O.; Ode, A.; Pfitzner, T.; Adjaye, J.; Kasper, G.; Duda, G.N. Functional comparison of chronological and in vitro aging: Differential role of the cytoskeleton and mitochondria in mesenchymal stromal cells. PLoS ONE 2012, 7, e52700. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.L.; Todhunter, M.E.; LaBarge, M.A.; Gartner, Z.J. Opportunities for organoids as new models of aging. J. Cell Biol. 2018, 217, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez Guimera, A.; Welsh, C.; Dalle Pezze, P.; Fullard, N.; Nelson, G.; Roger, M.F.; Przyborski, S.A.; Shanley, D.P. Systems modelling ageing: From single senescent cells to simple multi-cellular models. Essays Biochem. 2017, 61, 369–377. [Google Scholar] [PubMed]
- Wang, W.; Li, S.; Dong, H.P.; Lv, S.; Tang, Y.Y. Differential impairment of spatial and nonspatial cognition in a mouse model of brain aging. Life Sci. 2009, 85, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.M.; Kim, J.W.; Yoo, D.Y.; Yim, H.S.; Kim, D.W.; Choi, J.H.; Kim, W.; Jung, H.Y.; Won, M.H.; Hwang, I.K.; et al. Physical exercise ameliorates the reduction of neural stem cell, cell proliferation and neuroblast differentiation in senescent mice induced by D-galactose. BMC Neurosci. 2014, 15, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Liu, X.; Liu, J.; Li, H.; Yang, Y.; Liu, J.; Guo, Z.; Xiao, K.; Zhang, C.; Liu, J.; et al. D-galactose induces a mitochondrial complex I deficiency in mouse skeletal muscle: Potential benefits of nutrient combination in ameliorating muscle impairment. J. Med. Food 2014, 17, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Kovacic, P. Hydroxyurea (therapeutics and mechanism): Metabolism, carbamoyl nitroso, nitroxyl, radicals, cell signaling and clinical applications. Med. Hypotheses 2011, 76, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Su, X.; Liu, J.; Zhao, Y.; Li, Z.; Xu, X.; Li, H.; Nashun, B. Comparison of naturally aging and D-galactose induced aging model in beagle dogs. Exp. Ther. Med. 2017, 14, 5881–5888. [Google Scholar] [CrossRef] [Green Version]
- Bo-Htay, C.; Palee, S.; Apaijai, N.; Chattipakorn, S.C.; Chattipakorn, N. Effects of d-galactose-induced ageing on the heart and its potential interventions. J. Cell. Mol. Med. 2018, 22, 1392–1410. [Google Scholar] [CrossRef] [Green Version]
- Strait, J.B.; Lakatta, E.G. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail. Clin. 2012, 8, 143–164. [Google Scholar] [CrossRef] [Green Version]
- Sheydina, A.; Riordon, D.R.; Boheler, K.R. Molecular mechanisms of cardiomyocyte aging. Clin. Sci. 2011, 121, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Kanaporis, G.; Blatter, L.A. Alternans in atria: Mechanisms and clinical relevance. Medicina 2017, 53, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Cutler, M.J.; Wan, X.; Laurita, K.R.; Hajjar, R.J.; Rosenbaum, D.S. Targeted SERCA2a gene expression identifies molecular mechanism and therapeutic target for arrhythmogenic cardiac alternans. Circulation Arrhythmia Electrophysiol. 2009, 2, 686–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, S.M.; Bode, F.; Karasik, P.L.; Franz, M.R. Alternans of atrial action potentials during atrial flutter as a precursor to atrial fibrillation. Circulation 2002, 106, 1968–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, S.M.; Franz, M.R.; Clopton, P.; Pruvot, E.J.; Krummen, D.E. Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation 2011, 123, 2922–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morillo, C.A.; Klein, G.J.; Jones, D.L.; Guiraudon, C.M. Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation 1995, 91, 1588–1595. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Gupta, S.; Young, D.; Das, B.; McMahon, J.; Sen, S. Impairment of ultrastructure and cytoskeleton during progression of cardiac hypertrophy to heart failure. Lab. Investig. 2010, 90, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Furuoka, H.; Yagi, S.; Murakami, A.; Honma, A.; Kobayashi, Y.; Matsui, T.; Miyahara, K.; Taniyama, H. Hereditary dilated cardiomyopathy in Holstein-Friesian cattle in Japan: Association with hereditary myopathy of the diaphragmatic muscles. J. Comp. Pathol. 2001, 125, 159–165. [Google Scholar] [CrossRef]
- Feldman, M.L.; Navaratnam, V. Ultrastructural changes in atrial myocardium of the ageing rat. J. Anat. 1981, 133, 7–17. [Google Scholar]
- Pieri, C.; Recchioni, R.; Moroni, F.; Marcheselli, F.; Marra, M. Food restriction in female Wistar rats. VII. Mitochondrial parameters in resting and proliferating splenic lymphocytes. Arch. Gerontol. Geriatr. 1994, 19, 31–42. [Google Scholar] [CrossRef]
- Sugrue, M.M.; Wang, Y.; Rideout, H.J.; Chalmers-Redman, R.M.E.; Tatton, W.G. Reduced Mitochondrial Membrane Potential and Altered Responsiveness of a Mitochondrial Membrane Megachannel in p53-Induced Senescence. Biochem. Biophys. Res. Commun. 1999, 261, 123–130. [Google Scholar] [CrossRef]
- Niemann, B.; Chen, Y.; Teschner, M.; Li, L.; Silber, R.E.; Rohrbach, S. Obesity induces signs of premature cardiac aging in younger patients: The role of mitochondria. J. Am. Coll. Cardiol. 2011, 57, 577–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.K.; Cheng, C.C.; Tsai, M.C.; Wu, P.Y.; Chen, Y.A.; Chen, Y.C.; Chen, S.A.; Chen, Y.J. Mitochondrial dysfunction on sinoatrial node and pulmonary vein electrophysiological activities. Exp. Ther. Med. 2017, 13, 2486–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, L.L.; Li, W.; Lu, Y.; Centracchio, J.; Terentyeva, R.; Koren, G.; Terentyev, D. Redox modification of ryanodine receptors by mitochondria-derived reactive oxygen species contributes to aberrant Ca2+ handling in ageing rabbit hearts. J. Physiol. 2013, 591, 5895–5911. [Google Scholar] [CrossRef]
- Sag, C.M.; Santos, C.X.; Shah, A.M. Redox regulation of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2014, 73, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tribulova, N.; Szeiffova Bacova, B.; Benova, T.; Viczenczova, C. Can we protect from malignant arrhythmias by modulation of cardiac cell-to-cell coupling? J. Electrocardiol. 2015, 48, 434–440. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. The aging myocardium: Roles of mitochondrial damage and lysosomal degradation. Heartlung Circ. 2005, 14, 107–114. [Google Scholar] [CrossRef]
- Guo, X.; Yuan, S.; Liu, Z.; Fang, Q. Oxidation- and CaMKII-mediated sarcoplasmic reticulum Ca2+leak triggers atrial fibrillation in aging. J. Cardiovasc. Electrophysiol. 2014, 25, 645–652. [Google Scholar] [CrossRef]
- Huang, S.Y.; Chen, Y.C.; Kao, Y.H.; Hsieh, M.H.; Lin, Y.K.; Chen, S.A.; Chen, Y.J. Redox and Activation of Protein Kinase A Dysregulates Calcium Homeostasis in Pulmonary Vein Cardiomyocytes of Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6, e005701. [Google Scholar] [CrossRef]
- Terman, A.; Gustafsson, B.; Brunk, U.T. Autophagy, organelles and ageing. J. Pathol. 2007, 211, 134–143. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Antonucci, L.; Fagman, J.B.; Kim, J.Y.; Todoric, J.; Gukovsky, I.; Mackey, M.; Ellisman, M.H.; Karin, M. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6166–E6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meyer, G.R.; De Keulenaer, G.W.; Martinet, W. Role of autophagy in heart failure associated with aging. Heart Fail. Rev. 2010, 15, 423–430. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Henson, G.D.; Thorburn, A.; Sindler, A.L.; Pierce, G.L.; Seals, D.R. Translational evidence that impaired autophagy contributes to arterial ageing. J. Physiol. 2012, 590, 3305–3316. [Google Scholar] [CrossRef] [PubMed]
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; Zablocki, D.K.; Sadoshima, J. Aging and Autophagy in the Heart. Circ. Res. 2016, 118, 1563–1576. [Google Scholar] [CrossRef] [Green Version]
- Carroll, B.; Hewitt, G.; Korolchuk, V.I. Autophagy and ageing: Implications for age-related neurodegenerative diseases. Essays Biochem. 2013, 55, 119–131. [Google Scholar]
- Mozos, I. Laboratory markers of ventricular arrhythmia risk in renal failure. BioMed Res. Int. 2014, 2014, 509204. [Google Scholar] [CrossRef] [Green Version]
- Mozos, I.; Borzak, G.; Caraba, A.; Mihaescu, R. Arterial stiffness in hematologic malignancies. OncoTargets Ther. 2017, 10, 1381–1388. [Google Scholar] [CrossRef] [Green Version]
- Kälsch, H.; Wieneke, H.; Erbel, R. Acute myocardial infarction in a patient with chronic myelocytic leukemia during chemotherapy with hydroxyurea. Herz 2010, 35, 420–422. [Google Scholar] [CrossRef]
- Higazi, D.R.; Fearnley, C.J.; Drawnel, F.M.; Talasila, A.; Corps, E.M.; Ritter, O.; McDonald, F.; Mikoshiba, K.; Bootman, M.D.; Roderick, H.L. Endothelin-1-stimulated InsP3-induced Ca2+ release is a nexus for hypertrophic signaling in cardiac myocytes. Mol. Cell 2009, 33, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Medvedev, N.; Popov, V.; Henneberger, C.; Kraev, I.; Rusakov, D.A.; Stewart, M.G. Glia selectively approach synapses on thin dendritic spines. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014, 369, 20140047. [Google Scholar] [CrossRef] [Green Version]















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masoud, S.; McDonald, F.; Bister, D.; Kotecki, C.; Bootman, M.D.; Rietdorf, K. Examining Cardiomyocyte Dysfunction Using Acute Chemical Induction of an Ageing Phenotype. Int. J. Mol. Sci. 2020, 21, 197. https://doi.org/10.3390/ijms21010197
Masoud S, McDonald F, Bister D, Kotecki C, Bootman MD, Rietdorf K. Examining Cardiomyocyte Dysfunction Using Acute Chemical Induction of an Ageing Phenotype. International Journal of Molecular Sciences. 2020; 21(1):197. https://doi.org/10.3390/ijms21010197
Chicago/Turabian StyleMasoud, Said, Fraser McDonald, Dirk Bister, Claire Kotecki, Martin D. Bootman, and Katja Rietdorf. 2020. "Examining Cardiomyocyte Dysfunction Using Acute Chemical Induction of an Ageing Phenotype" International Journal of Molecular Sciences 21, no. 1: 197. https://doi.org/10.3390/ijms21010197
APA StyleMasoud, S., McDonald, F., Bister, D., Kotecki, C., Bootman, M. D., & Rietdorf, K. (2020). Examining Cardiomyocyte Dysfunction Using Acute Chemical Induction of an Ageing Phenotype. International Journal of Molecular Sciences, 21(1), 197. https://doi.org/10.3390/ijms21010197