METTL3 Regulates Osteoblast Differentiation and Inflammatory Response via Smad Signaling and MAPK Signaling

Abstract
:1. Introduction
2. Results
2.1. LPS Treatment Induces an Inflammatory Environment and Inhibits Osteoblast Differentiation
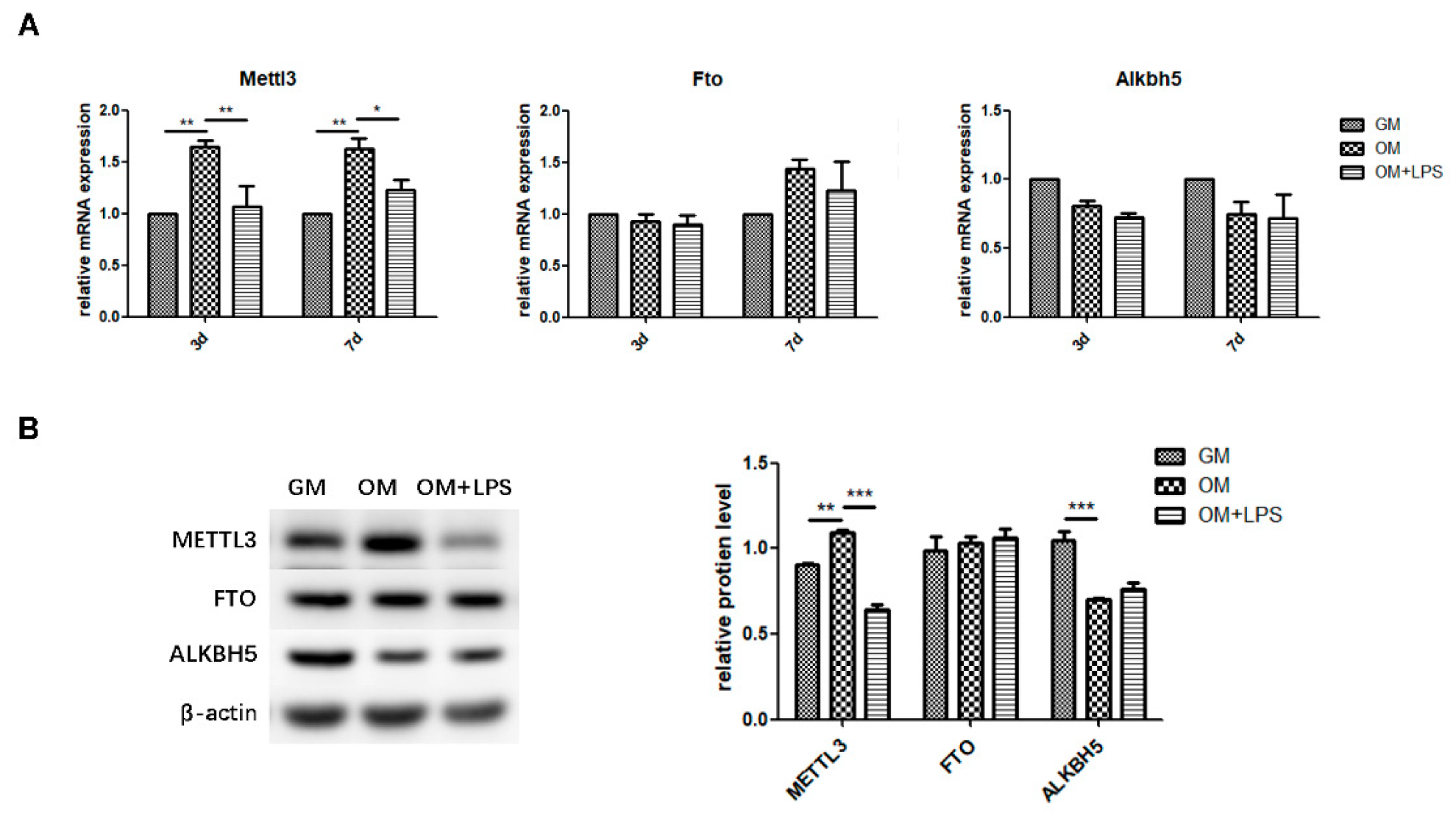
2.2. m6A Methyltransferase and Demethylase Expression during Osteogenesis and Inflammation
2.3. METTL3 Knockdown Inhibits Osteoblast Differentiation and Mineralization in LPS-Stimulated Osteoblasts
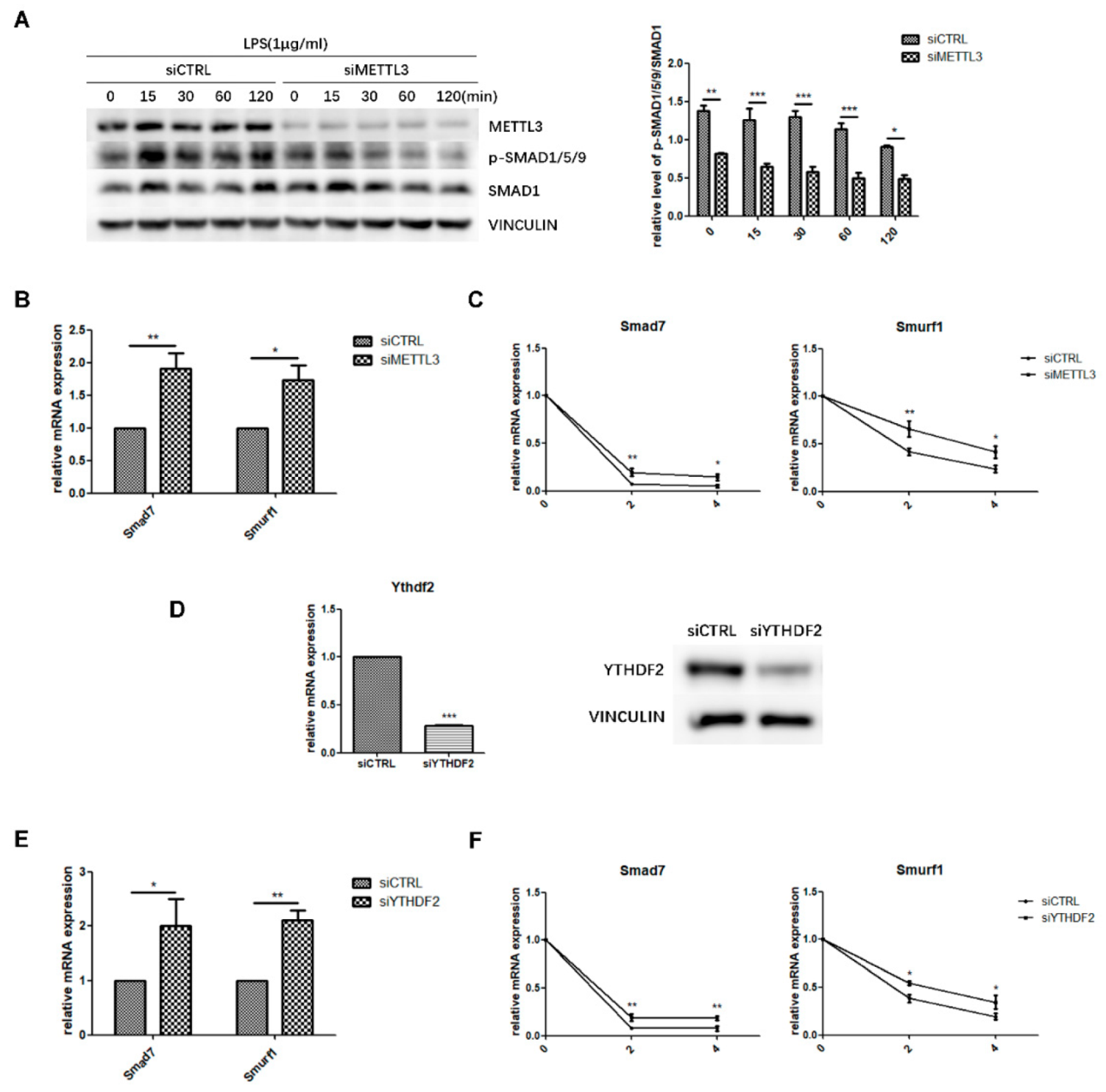
2.4. METTL3 May Affect Smad-Dependent Signaling by Regulating Smad7 and Smurf1 mRNA Stability Via YTHDF2 Involvement
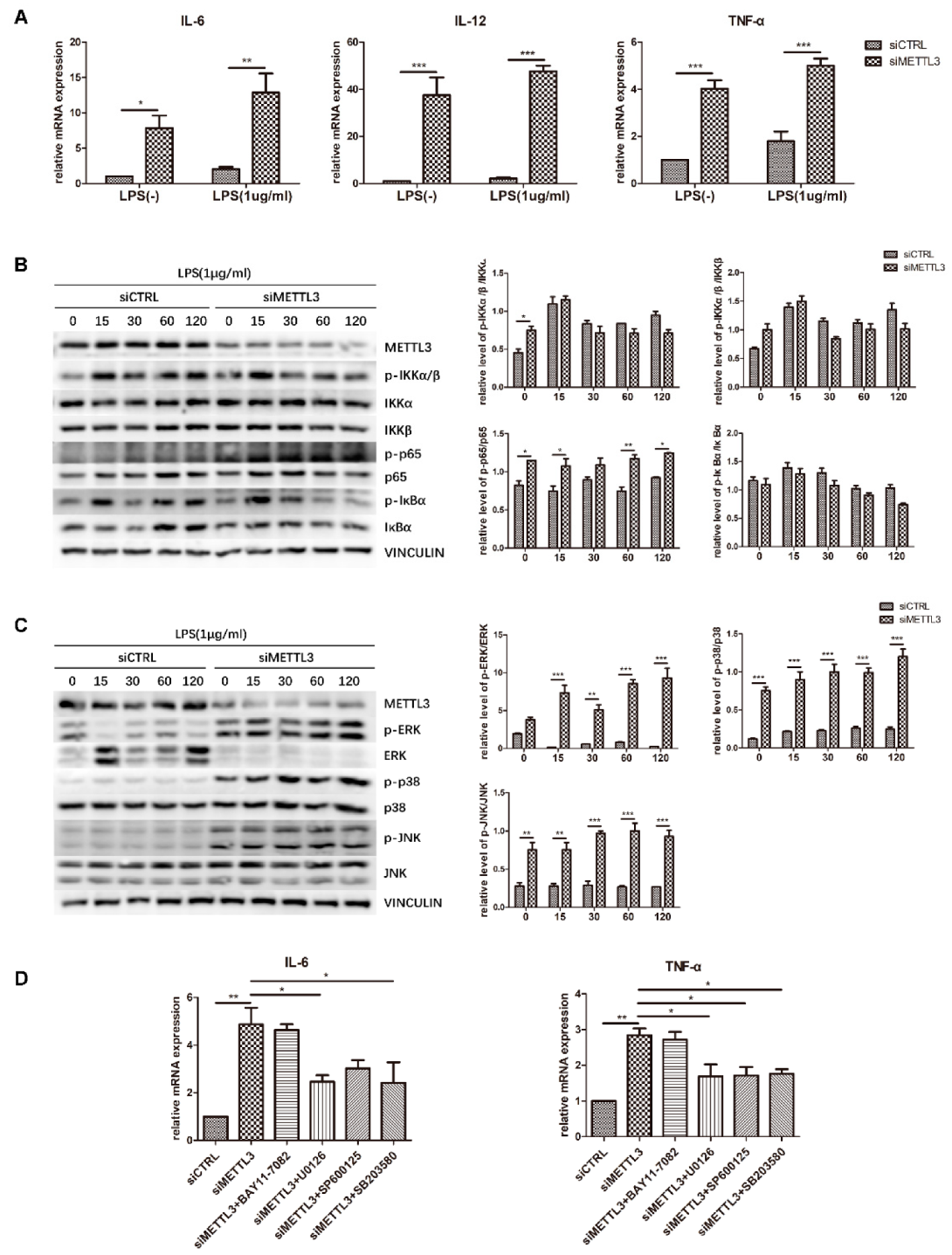
2.5. METTL3 Knockdown Activates MAPK Signaling to Promote Proinflammatory Cytokine Expression in LPS-Treated Osteoblasts
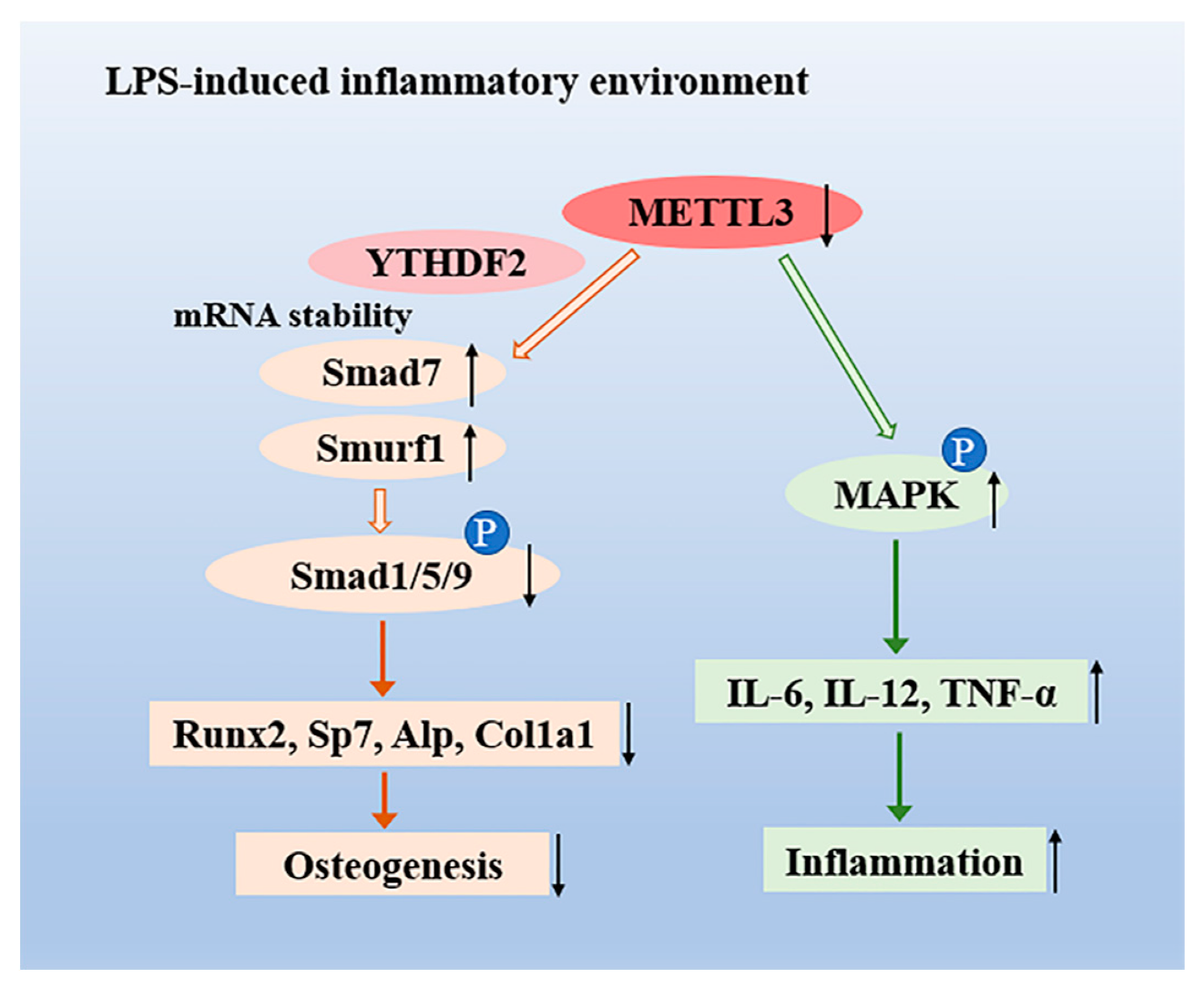
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Cell Transfection
4.3. Real-Time Quantitative Polymerase Chain Reaction (qRT-PCR)
4.4. Western Blotting Analysis
4.5. Alkaline Phosphatase and Alizarin Red Staining
4.6. mRNA Stability Measurement
4.7. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Long, F. Building strong bones: Molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol. 2011, 13, 27–38. [Google Scholar] [CrossRef]
- Maes, C.; Kobayashi, T.; Selig, M.K.; Torrekens, S.; Roth, S.I.; Mackem, S.; Carmeliet, G.; Kronenberg, H.M. Osteoblast Precursors, but Not Mature Osteoblasts, Move into Developing and Fractured Bones along with Invading Blood Vessels. Dev. Cell 2010, 19, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef]
- Wu, M.; Chen, G.; Li, Y. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef]
- Pacios, S.; Xiao, W.; Mattos, M.; Lim, J.; Tarapore, R.S.; Alsadun, S.; Yu, B.; Wang, C.; Graves, D.T. Osteoblast Lineage Cells Play an Essential Role in Periodontal Bone Loss Through Activation of Nuclear Factor-Kappa B. Sci. Rep. 2015, 5, 16694. [Google Scholar] [CrossRef]
- Josse, J.; Velard, F.; Gangloff, S.C. Staphylococcus aureus vs. Osteoblast: Relationship and Consequences in Osteomyelitis. Front. Cell. Infect. Microbiol. 2015, 5, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruotti, N.; Corrado, A.; Cantatore, F.P. Osteoblast role in osteoarthritis pathogenesis. J. Cell. Physiol. 2017, 232, 2957–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daigang, L.; Jining, Q.; Jinlai, L.; Pengfei, W.; Chuan, S.; Liangku, H.; Ding, T.; Zhe, S.; Wei, W.; Zhong, L.; et al. LPS-stimulated inflammation inhibits BMP-9-induced osteoblastic differentiation through crosstalk between BMP/MAPK and Smad signaling. Exp. Cell Res. 2016, 341, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Xing, Q.; Ye, Q.; Fan, M.; Zhou, Y.; Xu, Q.; Sandham, A. Porphyromonas gingivalis lipopolysaccharide inhibits the osteoblastic differentiation of preosteoblasts by activating Notch1 signaling. J. Cell. Physiol. 2010, 225, 106–114. [Google Scholar] [CrossRef]
- Tang, Y.; Zhou, X.; Gao, B.; Xu, X.; Sun, J.; Cheng, L.; Zhou, X.; Zheng, L. Modulation of Wnt/β-catenin Signaling Attenuates Periapical Bone Lesions. J. Dent. Res. 2013, 93, 175–182. [Google Scholar] [CrossRef]
- Kassem, A.; Henning, P.; Lundberg, P.; Souza, P.P.C.; Lindholm, C.; Lerner, U.H. Porphyromonas gingivalis Stimulates Bone Resorption by Enhancing RANKL (Receptor Activator of NF-κB Ligand) through Activation of Toll-like Receptor 2 in Osteoblasts. J. Biol. Chem. 2015, 290, 20147–20158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Pastou, T.C.; Gasque, P. Bone responses in health and infectious diseases: A focus on osteoblasts. J. Infect. 2017, 75, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of Methylated Nucleosides in Messenger RNA from Novikoff Hepatoma Cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, X.; Lu, Z.; Zhao, B.S.; Ma, H.; Hsu, P.J.; Liu, C.; He, C. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017, 27, 315–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N6-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.; Mumbach, M.R.; Jovanovic, M.; Wang, T.; Maciag, K.; Bushkin, G.G.; Mertins, P.; Ter-Ovanesyan, D.; Habib, N.; Cacchiarelli, D.; et al. Perturbation of m6A Writers Reveals Two Distinct Classes of mRNA Methylation at Internal and 5′ Sites. Cell Rep. 2014, 8, 284–296. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Doxtader, K.A.; Nam, Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 2016, 63, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.; Song, S.; et al. ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Gokhale, N.S.; McIntyre, A.B.R.; McFadden, M.J.; Roder, A.E.; Kennedy, E.M.; Gandara, J.A.; Hopcraft, S.E.; Quicke, K.M.; Vazquez, C.; Willer, J.; et al. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 2016, 20, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Bi, Z.; Wu, R.; Zhao, Y.; Liu, Y.; Liu, Q.; Wang, Y.; Wang, X. METTL3 inhibits BMSC adipogenic differentiation by targeting the JAK1/STAT5/C/EBPβ pathway via an m6A-YTHDF2–dependent manner. FASEB J. 2019, 33, 7529–7544. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Chai, G.; Wu, Y.; Li, J.; Chen, F.; Liu, J.; Luo, G.; Tauler, J.; Du, J.; Lin, S.; et al. RNA m6A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat. Commun. 2019, 10, 2065. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Lin, Z.; Wan, A.; Chen, H.; Liang, H.; Sun, L.; Wang, Y.; Li, X.; Xiong, X.; Wei, B.; et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol. Caner 2019, 18, 46. [Google Scholar] [CrossRef] [Green Version]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef]
- Wu, Y.; Xie, L.; Wang, M.; Xiong, Q.; Guo, Y.; Liang, Y.; Li, J.; Sheng, R.; Deng, P.; Wang, Y.; et al. Mettl3-mediated m6A RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nat. Commun. 2018, 9, 4772. [Google Scholar] [CrossRef]
- Tian, C.; Huang, Y.; Li, Q.; Feng, Z.; Xu, Q. Mettl3 Regulates Osteogenic Differentiation and Alternative Splicing of Vegfa in Bone Marrow Mesenchymal Stem Cells. Int. J. Mol. Sci. 2019, 20, 551. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Zhao, B.S.; Wang, X.; Beadell, A.V.; Lu, Z.; Shi, H.; Kuuspalu, A.; Ho, R.K.; He, C. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 2017, 542, 475–478. [Google Scholar] [CrossRef] [Green Version]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholler, E.; Weichmann, F.; Treiber, T.; Ringle, S.; Treiber, N.; Flatley, A.; Feederle, R.; Bruckmann, A.; Meister, G. Interactions, localization, and phosphorylation of the m(6)A generating METTL3-METTL14-WTAP complex. RNA 2018, 24, 499–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, K.D.; Jaffrey, S.R. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 2014, 15, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Zhao, Y.; He, J.; Zhang, Y.; Xi, H.; Liu, M.; Ma, J.; Wu, L. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4–NOT deadenylase complex. Nat. Commun. 2016, 7, 12626. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Li, Q.; Meng, R.; Yi, B.; Xu, Q. METTL3 regulates alternative splicing of MyD88 upon the lipopolysaccharide-induced inflammatory response in human dental pulp cells. J. Cell. Mol. Med. 2018, 22, 2558–2568. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef]
- Li, H.; Tong, J.; Zhu, S.; Batista, P.J.; Duffy, E.E.; Zhao, J.; Bailis, W.; Cao, G.; Kroehling, L.; Chen, Y.; et al. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 2017, 548, 338–342. [Google Scholar] [CrossRef] [Green Version]
- Winkler, R.; Gillis, E.; Lasman, L.; Safra, M.; Geula, S.; Soyris, C.; Nachshon, A.; Tai-Schmiedel, J.; Friedman, N.; Le-Trilling, V.T.K.; et al. m6A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 2019, 20, 173–182. [Google Scholar] [CrossRef]
- Florencio-Silva, R.; Sasso, G.R.D.S.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed. Res. Int. 2015, 2015, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Bandow, K.; Maeda, A.; Kakimoto, K.; Kusuyama, J.; Shamoto, M.; Ohnishi, T.; Matsuguchi, T. Molecular mechanisms of the inhibitory effect of lipopolysaccharide (LPS) on osteoblast differentiation. Biochem. Biophys. Res. Commun. 2010, 402, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.D.; Naylor, A.J.; Buckley, C.; Filer, A.; Tak, P.P. Fibroblasts and Osteoblasts in Inflammation and Bone Damage. Adv. Exp. Med. Biol. 2018, 1060, 37–54. [Google Scholar] [PubMed]
- Guo, C.; Yuan, L.; Wang, J.; Wang, F.; Yang, X.; Zhang, F.; Song, J.; Ma, X.; Cheng, Q.; Song, G. Lipopolysaccharide (LPS) Induces the Apoptosis and Inhibits Osteoblast Differentiation Through JNK Pathway in MC3T3-E1 Cells. Inflammation 2014, 37, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Baum, R.; Gravallese, E.M. Impact of Inflammation on the Osteoblast in Rheumatic Diseases. Curr. Osteoporos. Rep. 2014, 12, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Rottman, F.M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997, 3, 1233–1247. [Google Scholar]
- Chen, G.; Deng, C.; Li, Y. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Liu, Z.; Chen, Y. Regulation of TGF-signaling by Smad7. Acta Bioch. Biophys. Sin. 2009, 41, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Xie, Z.; Ma, Y.; Pan, X.; Wang, J.; Chen, Z.; Shi, P. TGF-β inhibits osteogenesis by upregulating the expression of ubiquitin ligase SMURF1 via MAPK-ERK signaling. J. Cell. Physiol. 2018, 233, 596–606. [Google Scholar] [CrossRef]
- Wang, H.; Zuo, H.; Liu, J.; Wen, F.; Gao, Y.; Zhu, X.; Liu, B.; Xiao, F.; Wang, W.; Huang, G.; et al. Loss of YTHDF2-mediated m(6)A-dependent mRNA clearance facilitates hematopoietic stem cell regeneration. Cell Res. 2018, 28, 1035–1038. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Wei, L.; Law, C.T.; Tsang, F.H.C.; Shen, J.; Cheng, C.L.H.; Tsang, L.H.; Ho, D.W.H.; Chiu, D.K.C.; Lee, J.M.F.; et al. RNA N6-Methyladenosine Methyltransferase-Like 3 Promotes Liver Cancer Progression Through YTHDF2-Dependent Posttranscriptional Silencing of SOCS2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef]
- Nagao, M.; Tanabe, N.; Manaka, S.; Naito, M.; Sekino, J.; Takayama, T.; Kawato, T.; Torigoe, G.; Kato, S.; Tsukune, N.; et al. LIPUS suppressed LPS-induced IL-1α through the inhibition of NF-κB nuclear translocation via AT1-PLCβ pathway in MC3T3-E1 cells. J. Cell. Physiol. 2017, 232, 3337–3346. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| siRNA | Sequences (5′-3′) |
|---|---|
| METTL3 siRNA #1 | CAGCUCAGGAGUUGAUUGAGGUAAA |
| UUUACCUCAAUCAACUCCUGAGCUG | |
| METTL3 siRNA #2 | UCGUUAGUCUCUGGUCUGAACUCUU |
| AAGAGUUCAGACCAGAGACUAACGA | |
| YTHDF2 siRNA | CCAUGAUUGAUGGACAGUCAGCUUU |
| AAAGCUGACUGUCCAUCAAUCAUGG |
| Gene | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| IL6 | TCTATACCACTTCACAAGTCGGA | GAATTGCCATTGCACAACTCTTT |
| IL12 | GTCCTCAGAAGCTAACCATCTCC | CCAGAGCCTATGACTCCATGTC |
| TNF-a | CCACCACGCTCTTCTGTCTA | GGTCTGGGCCATAGAACTGA |
| Runx2 | TTCAACGATCTGAGATTTGTGGG | GGATGAGGAATGCGCCCTA |
| Sp7 | ATGGCGTCCTCTCTGCTTG | TGAAAGGTCAGCGTATGGCTT |
| Alpl | GGCTGGAGATGGACAAATTCC | CCGAGTGGTAGTCACAATGCC |
| Col1a1 | CCCTGCCTGCTTCGTGTA | TTGAGTTTGGGTTGTTCGTC |
| Mettl3 | CTTTCTACCCCATCTTGAGTG | CCAACCTTCCGTAGTGATAGTC |
| Fto | GACTCGTCCTCACTTTCATCC | AAGAGCAGAGCAGCCTACAAC |
| Alkbh5 | GTGGGACCTTTTGGGTTTCAG | GCATACGGCCTCAGGACATTA |
| Ythdf2 | ATAGGAAAAGCCAATGGAGGG | CCAAAAGGTCAAGGAAACAAAG |
| Smad7 | GGGCTTTCAGATTCCCAACTT | AGGGCTCTTGGACACAGTAGA |
| Smurf1 | ACACTGGCTACCAGCGTTTG | TCTGTCTCGGGTCTGTAAACTG |
| Gapdh | GGTCATCCCAGAGCTGAACG | TGCTGTTGAAGTCGCAGGA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Gu, X.; Li, D.; Cai, L.; Xu, Q. METTL3 Regulates Osteoblast Differentiation and Inflammatory Response via Smad Signaling and MAPK Signaling. Int. J. Mol. Sci. 2020, 21, 199. https://doi.org/10.3390/ijms21010199
Zhang Y, Gu X, Li D, Cai L, Xu Q. METTL3 Regulates Osteoblast Differentiation and Inflammatory Response via Smad Signaling and MAPK Signaling. International Journal of Molecular Sciences. 2020; 21(1):199. https://doi.org/10.3390/ijms21010199
Chicago/Turabian StyleZhang, Yiwen, Xiaofei Gu, Di Li, Luhui Cai, and Qiong Xu. 2020. "METTL3 Regulates Osteoblast Differentiation and Inflammatory Response via Smad Signaling and MAPK Signaling" International Journal of Molecular Sciences 21, no. 1: 199. https://doi.org/10.3390/ijms21010199
APA StyleZhang, Y., Gu, X., Li, D., Cai, L., & Xu, Q. (2020). METTL3 Regulates Osteoblast Differentiation and Inflammatory Response via Smad Signaling and MAPK Signaling. International Journal of Molecular Sciences, 21(1), 199. https://doi.org/10.3390/ijms21010199