1. Introduction
Selective barriers formed by epithelial monolayers are vital for many critical physiological processes [
1]. These barriers are composed of intercellular multiprotein complexes along the apical-basal axis of epithelial cells creating direct cell-cell interactions and forming apical junctional complexes [
2]. These complexes include tight junctions (TJs), adherens junctions, and desmosomes.
TJs are the most apical and diverse of these junctional complexes. They are formed by transmembrane as well as cytoplasmic proteins and are linked to the cytoskeleton. The protein composition of TJs varies greatly among different epithelia and determines the barrier properties and permeability [
3].
Overall, TJs serve a dual function in epithelial layers acting as (a) a fence, separating membrane proteins between the apical and basolateral membrane and (b) as a gate by regulating size- and charge-selective movements of ions, solutes, and small molecules via the paracellular route [
4,
5,
6,
7]. Consequently, TJs are not only critical to establish and maintain cell adhesion and epithelial polarity but are also necessary to create selective paracellular permeability between compartments.
Several studies show that the main protein family mediating TJ characteristics and functions are the membrane-spanning claudins. So far, more than 25 different claudins have been identified, and at least 10 of them are expressed in spatiotemporal patterns along the renal nephron (for a detailed review see [
8]).
Structurally, the transmembrane proteins connect in both
cis and
trans to neighboring claudins of the same, as well as the opposing, cell membranes forming TJ strands [
9]. Claudins are sufficient to arrange those strands when expressed in cells lacking endogenous TJ formation [
10].
Here, we review the role of claudins in epithelial barrier formation with emphasis on the renal collecting duct as well as their implications in collecting duct physiology and function.
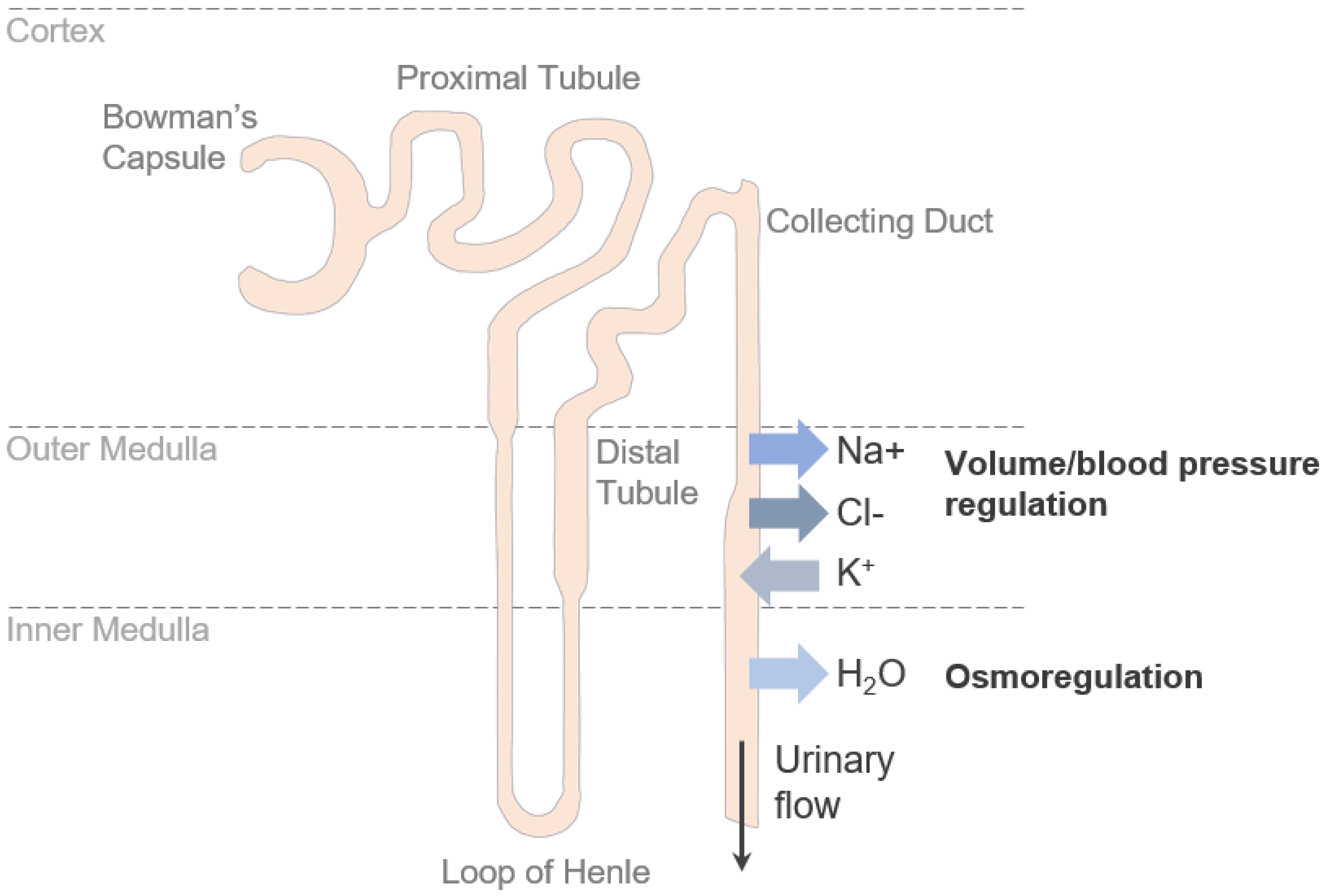
2. The Renal Collecting Duct
The renal collecting duct is the most distal part of the renal tubules. It connects renal nephrons with the renal pelvis and—together with the distal convoluted tubule and the connecting tubule—it contributes to the aldosterone-sensitive distal nephron. In general, the renal collecting duct plays important roles in fine-tuning urinary composition, extracellular fluid volume, electrolyte balance, blood pressure regulation, water homeostasis, and acid-base regulation [
11,
12].
Although most of the water and solute reabsorption in the kidney occurs in the more upstream segments of the nephron, transport variability in the renal collecting duct is significantly higher, and ion and water transport are under strict hormonal control [
11]. This permits to adjust reabsorption and secretion to prevalent physiological conditions and to control the body’s water and electrolyte balance closely.
The main solutes reabsorbed in the collecting duct are sodium (Na+) and chloride (Cl−), while potassium (K+) is secreted into the urine [
13] (
Figure 1). Transport of these ions occurs via the transcellular route—mediated by channels and membrane transporters—and via the paracellular route—mediated by TJs.
3. Claudins and Their Roles in Collecting Duct Epithelial Barrier Formation and Paracellular Ion Transport
In general, claudins can be pore- or barrier-forming [
14]. As in other segments of the renal nephron, it is believed that the expressed claudins are the main mediators of TJ permeability characteristics, and thus, control the paracellular pathway in the collecting duct [
9,
15]. Among the predominately expressed claudins in the renal collecting duct are claudin-3 (Cldn-3), -4 (Cldn-4), -7 (Cldn-7) and -8 (Cldn-8) [
16,
17]. Furthermore, a recently published study proposes a role for claudin-19 (Cldn-19) in the renal collecting duct [
18]. Overall, the present knowledge strongly supports the theory that these claudins either enforce the epithelial barrier in general or build a paracellular Cl− channel, while other aspects of their functions are less clear (
Table 1).
In the following sections, we will briefly review the published literature on the functions of these claudins in the collecting duct. As a general note, it should be acknowledged that gain-of-function studies utilizing overexpression of claudins in cell lines have a general limitation due to the influence of the endogenous components of the TJ in each cell line. The expressed claudins in the respective chosen model make it difficult to determine the exact impact of single claudins on permeability characteristics. In contrast, loss-of-function studies, if carried out in the appropriate system (i.e., the collecting duct in vivo or cell lines that accurately represent collecting duct epithelia), are more appropriate to outline the molecular functions of each claudin.
3.1. Cldn-3
Several studies have found that Cldn-3 enforces the paracellular barrier [
19,
20]. Its overexpression in otherwise leaky Madin-Darby Canine Kidney (MDCK) II cells leads to an altered TJ structure with increased transepithelial resistance (TER) and decreased permeability for ions as well as molecules of 332 Da and 4 kDa, indicating a general role for Cldn-3 in barrier formation and enforcement [
19]. Furthermore, Cldn-3 has been described to promote tubule formation in vitro and might thus play a role during tubule morphogenesis in the developing kidney [
30]. The conditional knockout of Cldn-3 resulted in increased urinary pH without any effect on urine or plasma electrolytes [
21]. However, the collecting duct-specific role of Cldn-3 has not been studied so far.
3.2. Cldn-7
Cldn-7-deficient mice die shortly after birth due to severe renal salt wasting and dehydration [
27]. Interestingly, collecting duct cells isolated from Cldn-7-deficient mice demonstrate an increase in TER and show decreased paracellular permeability for both Na+ and Cl− [
31]. This suggests that Cldn-7 may form a non-selective paracellular channel facilitating Cl− and Na+ reabsorption in collecting ducts.
Different and partially conflicting roles of Cldn-7 have been suggested in other experimental systems, complicating the interpretation of Cldn-7 biology. Overexpression of Cldn-7 in LLC-PK1 cells induces an increase in TER accompanied by a reduced Cl− and elevated Na+ conductance [
24]. In contrast, the knockdown of Cldn-7 in those cells leads to loss of anion selectivity and decreased Cl− permeation, whereas in cation-selective MDCK cells, knockdown of Cldn-7 leads to increased Na+ permeability [
25]. Mutation experiments in vitro demonstrated the importance of specific negatively charged amino acids in the extracellular loop of Cldn-7 for paracellular permeability and charge selectivity [
32]. These contrasting results could indicate different roles for Cldn-7, depending on the cellular background, and endogenously expressed TJ components.
3.3. Cldn-4 and Cldn-8
Both, Cldn-4 and Cldn-8, are thought to serve as a cation barrier [
33,
34,
35] and an anion channel [
22]. This hypothesis is supported by data from in vivo experiments. Collecting duct-specific knockout of either Cldn-4 or Cldn-8, causes hypotension, hypochloremia, metabolic alkalosis and renal wasting of Na+ and Cl− [
23,
29]. These phenotypes would be consistent with Cldn-4 and Cldn-8 acting as paracellular Cl− channels, which are necessary for a paracellular “chloride shunt” required for effective transcellular Na+ reabsorption (via the epithelial sodium channel).
Different effects of Cldn-4 overexpression on charge selectivity, depending on the used cell model, have been described. Overexpression of Cldn-4 leads to an increase in TER in cation-selective MDCK II and anion-selective LLC-PK1 cells, but a decrease in Na+ permeability is only observed in MDCK II cells [
34].
Cldn-8 has been shown to be necessary to recruit Cldn-4 to the TJ and to implement the protein into the junctional complex. In the absence of Cldn-8, Cldn-4 is mainly found in the endoplasmic reticulum and the Golgi complex, but not in the apical cell membrane where TJs are located [
22]. Thus, Cldn-8 knockout causes a functional double knockout on the TJ level due to its requirement for correct Cldn-4 localization [
22].
3.4. Cldn-19
In addition to the longer known claudins of the renal collecting duct described above, a recently published study indicates a role for Cldn-19, which had previously been linked to thick ascending limb functions [
36,
37,
38]. Cldn-19 is associated with tightness and cation selectivity of the epithelial barrier. Interestingly, the TJ localization of Cldn-19 is promoted by a low osmolality, whereas high osmolality favors an intracellular localization, suggesting that it may contribute to tonicity-induced changes in paracellular ion selectivity [
18]. This role of a claudin in epithelial adaption to the changing osmolality along the corticomedullary axis provides an interesting aspect of TJ physiology but collecting duct-specific knockout models of Cldn-19 have not yet been generated.
4. The Collecting Duct Epithelial Barrier in Electrolyte-Free Water Reabsorption
Although water transport in the renal collecting duct is not directly facilitated by water channel-forming claudins (contrasting with observations of Cldn-2 in the proximal tubule [
39]), TJs in the collecting duct contribute to water reabsorption indirectly.
The driving force for water reabsorption in the renal collecting duct is a steep osmolality gradient formed by high concentrations of osmolytes in the interstitium, which increases towards the renal medulla [
40]. The tight collecting duct epithelial barrier is crucial to maintain the osmolality gradient between the tubular lumen and the interstitium.
Water transport in the renal collecting duct is mainly controlled by arginine vasopressin (AVP), also called antidiuretic hormone. If the water content in the body is low, AVP binds to its type 2 receptor (V2R) localized in the basal cell membrane of collecting duct principal cells and stimulates the expression of aquaporin-2 (AQP2) water channels. Furthermore, AVP triggers a signaling cascade leading to the accumulation of AQP2 in the apical membrane (for a detailed review see [
11,
41]). This mechanism enables reabsorption of electrolyte-free water from the intraluminal urine and forms the basis of urinary concentrating ability. Inactivation of either V2R or AQP2 leads to polyuria with massive excretion of electrolyte-free water, a condition called nephrogenic diabetes insipidus [
42].
A recent study has demonstrated the importance of an intact epithelial barrier in the renal collecting duct for efficient water reabsorption [
43]. Deletion of the transcriptional regulator Grainyhead-like 2 (Grhl2), an epithelial transcription factor that induces the expression of barrier-enforcing molecular TJ components including Cldn-4 [
44], results in a leaky collecting duct epithelium and a decreased TER across the collecting duct epithelium. Leakage of interstitial osmolytes across the Grhl2-deficient collecting duct epithelium is associated with defective retention of osmolytes in the interstitium of the inner medulla. Grhl2-deficient mice show signs of diabetes insipidus and fail to concentrate their urine adequately, although AQP-mediated water transport across the apical and basolateral membranes of Grhl2-deficient collecting ducts is intact [
43]. This indicates that a tight collecting duct epithelial barrier is crucial for the maintenance of osmolality gradients and for effective collecting duct water reabsorption. Interestingly, Grhl2 deficiency (unlike deficiencies of individual claudins) was not associated with abnormalities of Cl− and Na+ reabsorption. It needs to be acknowledged that Grhl2 functions are not restricted to the effects on the TJ, which may explain the difference in phenotypes. In addition, it is possible that the barrier defect of Grhl2-deficient collecting duct cells is more profound, leading to non-ion-selective leakage of interstitial osmolytes into hypotonic urine.
5. Aldosterone and Its Role in Transcellular and Paracellular Transport Regulation
In the renal collecting duct, Na+ transport is separated from Cl− transport [
45]. Na+ reabsorption from the intraluminal urine occurs transcellularly via the epithelial sodium channel (ENaC) that locates to the apical membrane of collecting duct principal cells. This generates a lumen-negative potential, providing a driving force for K+ secretion via the renal outer medullary potassium channel (ROMK). In contrast, Cl− reabsorption in the collecting duct occurs predominantly via the paracellular route. This “chloride shunt” is important to limit the built-up of a lumen-negative potential and, thereby, facilitates continued Na+ reabsorption and prevents excessive K+ secretion.
The key regulator of ENaC is aldosterone, a hormone secreted from the adrenal gland in response to hyperkalemia and hypovolemia [
11,
46]. Overall, aldosterone plays a central role in blood pressure regulation by controlling plasma Na+ and K+ levels and thus indirectly influences water retention or loss. However, growing evidence supports the hypothesis that aldosterone controls Na+ reabsorption and K+ secretion not only by regulating the abundance of ENaC and increasing transcellular transport but also by adjusting paracellular Cl− permeability in multiple ways:
For instance, aldosterone triggers the expression of Cldn-8 when ENaC is active, presumably to seal the paracellular route for Na+ back flux. Thereby net flux of Na+ can be increased [
28].
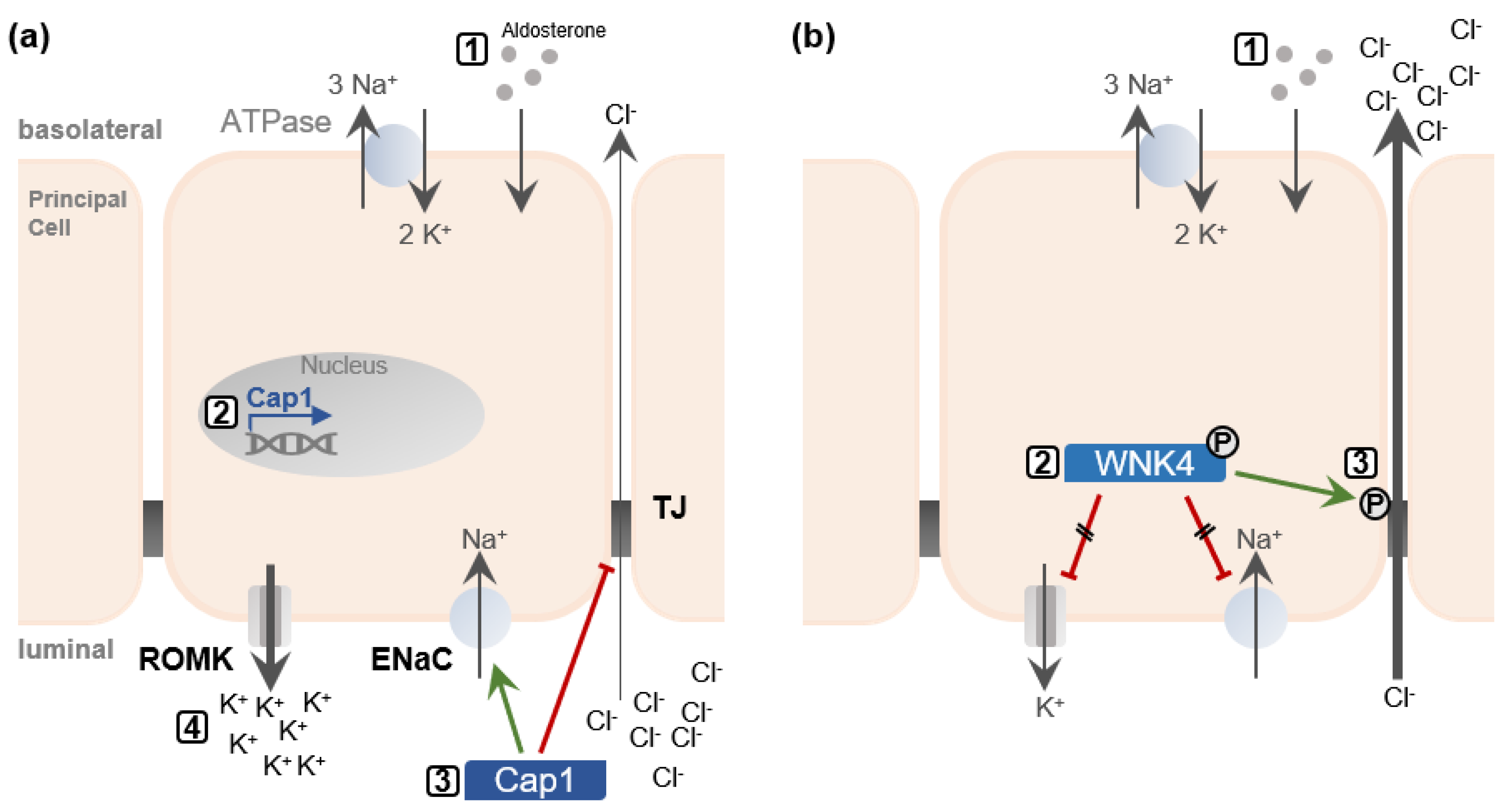
Additionally, aldosterone regulates channel-activating protease 1 (Cap1) [
47]. Cap1, in turn, stimulates ENaC and inhibits the Cl− conductivity by directly regulating Cldn-4
trans-interactions [
23,
48]. Thereby K+ secretion is favored over Cl− reabsorption (
Figure 2a).
Aldosterone also induces the phosphorylation and activation of the with no lysine kinases 4 (WNK4) [
49]. Expression of WNK4 in MDCK II cells has been shown to reduce the TER and increase Cl− permeability, without affecting TJ structure [
50]. Activated WNK4 phosphorylates Cldn-4 on threonine residues decreasing the cells’ TER and increasing apical to basal anion passage [
51].
In the absence of aldosterone, WNK4 inhibits ENaC and ROMK activity and thus directly opposes Cap1 [
52,
53]. WNK4 phosphorylation suspends this inhibition (
Figure 2b). Taken together, this indicates the possibility that aldosterone might regulate claudins through Cap1 and WNK4, coordinating Cl− reabsorption or K+ secretion, respectively.
Kahle and colleagues proposed WNK4 as the functional switch regulating Na+ and Cl− reabsorption independently from K+ secretion depending on the physiological conditions [
53]. However, the factors facilitating different functional states of WNK4 haven’t been provided, and to our knowledge, it is still unknown how the reverse actions of Cap1 and WNK4, that are both mediated by aldosterone, are regulated to decide in favor of K+ secretion or Cl− reabsorption, respectively.
In diabetes, the role of aberrant aldosterone signaling in the progression of renal disease has long been established [
54,
55]. Mediated by the divergent aldosterone levels, Cldn-4 and Cldn-8 are overexpressed in the distal nephron from type 1 diabetic rats, and the expression of WNK4 and its co-localization with Cldn-4 and Cldn-8 is also increased [
56,
57]. This might result in increased activation of Cldn-4 and Cldn-8 by WNK4 under diabetic conditions and could implicate disturbed paracellular transport in renal disease progression. However, additional experimental evidence verifying this hypothesis is needed.
6. Chloride Reabsorption in Renal Collecting Ducts and Potential Involvement in Disease
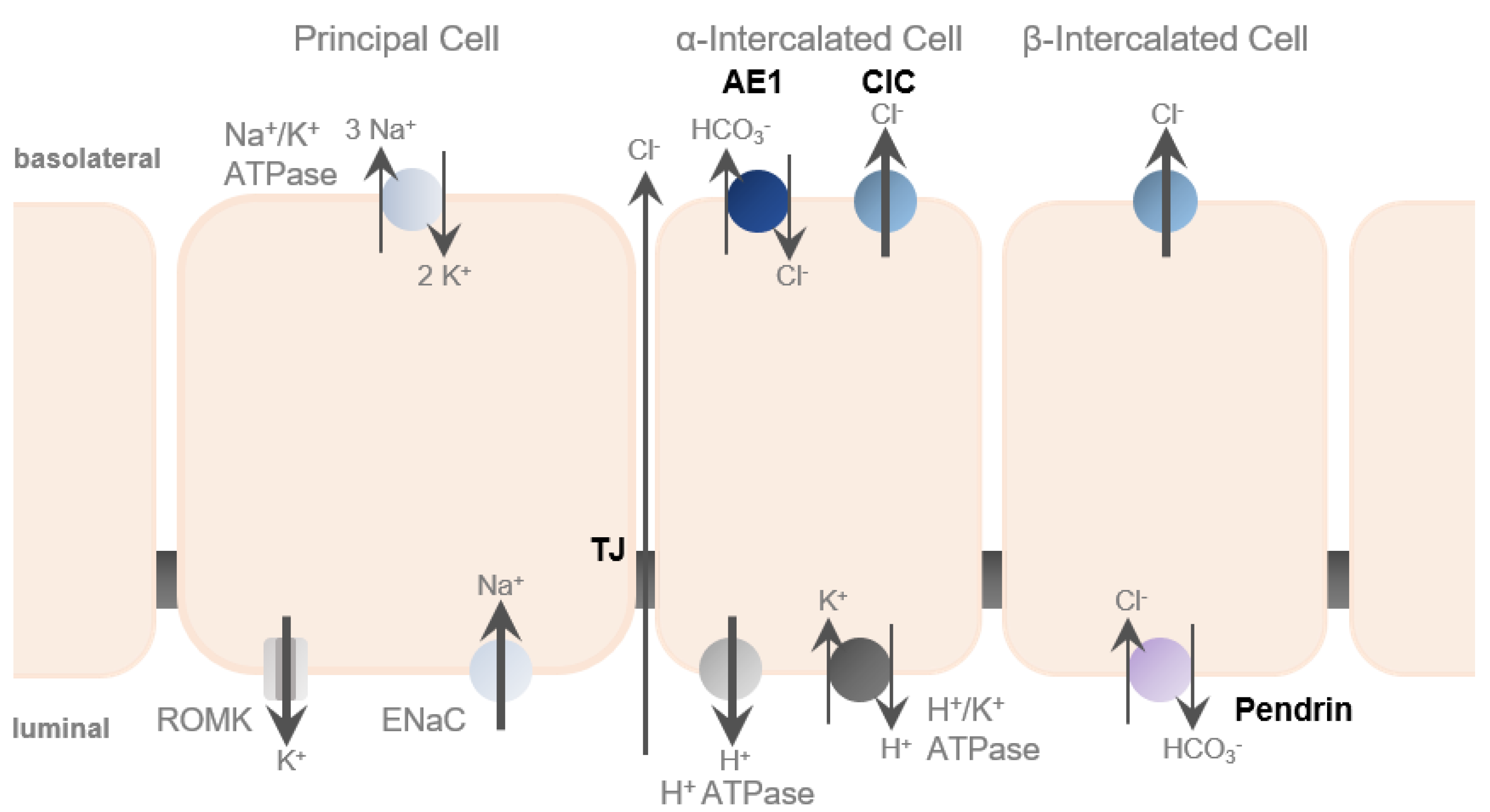
The aldosterone-sensitive distal nephron is the main side of Cl− reabsorption. It occurs via multiple ways, including paracellular transport via TJs as well as transcellular pathways in intercalated cells [
12,
23,
29] and is driven by the lumen-negative transepithelial potential generated by the unilateral Na+ reabsorption to maintain electroneutrality (
Figure 3). As in aldosterone signaling, it becomes increasingly evident that crosstalk between paracellular and transcellular transport occurs.
The importance of efficient Cl− reabsorption becomes obvious in claudin-deficient mouse models. For example, Cldn-7 deficiency in mice is lethal within 12 days after birth due to severe salt-wasting and subsequent chronic dehydration (see above) [
27]. Cldn-7
−/− mice show reduced ROMK and increased ENaC, Aqp2, and Na+ Cl− cotransporter mRNA. These changes in channel expressions are probably due to a compensatory mechanism to inhibit further urinary loss of salt and water [
27].
It has long been established that accumulation of luminal Cl− depolarizes the membrane, and thereby, inhibits the apical Na+ channel ENaC [
58]. Several studies also have demonstrated that pathological increases in Cl− reabsorption are associated with diseases, such as pseudo-hypoaldosteronism type II (PHA-II) or Gordon’s syndrome.
PHA-II is a rare Mendelian syndrome leading to hypertension, hyperkalemia, and metabolic acidosis [
59]. These symptoms are the exact opposite of the Cldn-4 and Cldn-8 knockout phenotypes in mice [
23,
29].
Interestingly, a long-known causative gene for PHA-II is WNK4 [
60], and WNK4 can regulate Cl− conductance presumably by phosphorylation of Cldn-4 and Cldn-8 [
50]. The PHA-II-causing mutation of WNK4 increases paracellular Cl− permeability in vitro [
49,
50]. Consistent with the proposed Cl− pore activity of Cldn-4 and Cldn-8, this is an additional indicator of an activating effect of claudin phosphorylation by WNK4. Furthermore, a recent study demonstrates that WNK4 is overexpressed in Cldn-7-deficient cultured, collecting duct cells [
31]. Conversely, the PHA-II mutant of WNK4 is associated with increased Cldn-7 phosphorylation and enhanced paracellular Cl− conductivity [
26]. This adds a new aspect and raises the possibility that claudins might act both up- and down-stream of WNK4 to regulate paracellular transport.
Another gene associated with PHA-II is KLHL3, encoding Kelch-like protein 3 [
61,
62]. Interestingly, KLHL3 normally induces ubiquitination and degradation of Cldn-8, while disease-associated mutations of KLHL3 abolish the interaction of KLHL3 with Cldn-8 [
29]. In addition, KLHL3 leads to the ubiquitination of WNK4. In line with PHA-II symptoms, loss of KLHL3 increases Cl- permeability in vivo [
29], which may contribute to the disease phenotype.
7. Conclusions
Collecting duct epithelia express claudins enforcing a high TER such as Cldn-3, -4, -7, -8, and -19, consistent with a demand for strong epithelial barrier function in the presence of steep transepithelial gradients. Based on our current knowledge, these claudins are thought to either support a tight barrier in general or to act as a cation barrier and/or an anion channel.
Consequently, the renal collecting duct is comprised of an especially tight epithelial barrier compared to other, more upstream segments of the nephron. As a result, the collecting duct lumen and interstitium are strictly separated. Nevertheless, the TJs of the collecting duct exhibit a regulated paracellular permeability for ions such as Cl-. Hence, the collecting duct TJ participates in a range of physiological functions:
It allows for controlled paracellular transport of Cl- to the interstitium, which is important in the setting of Na+ reabsorption via ENaC and secretion of K+ via ROMK.
It prevents paracellular diffusion and back flux of osmolytes into the urine and thus promotes the formation of steep gradients across the epithelium, which are necessary to drive electrolyte-free water reabsorption.
Future research will be needed to elucidate further the precise mechanisms that regulate TJ properties and that mediate crosstalk between paracellular and transcellular transport processes and how these processes relate to renal pathophysiology.
Author Contributions
Writing—original draft preparation: J.L., K.M.S.-O.; Writing—review and editing: J.L., K.M.S.-O. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the German Research Foundation (DFG) Research Training Group GRK 2318, by Collaborative Research Grant 1365, and by Research Unit FOR 2841. We acknowledge support from the German Research Foundation (DFG) and the Open Access Publication Fund of Charité-Universitätsmedizin Berlin.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AQP | Aquaporin |
| AVP | Arginine vasopressin |
| Cap1 | Channel-activating protease 1 |
| Cldn | Claudin |
| Cl- | Chloride |
| ENaC | Epithelial sodium channel |
| Grhl2 | Grainyhead-like 2 |
| K+ | Potassium |
| KLHL3 | Kelch-like protein 3 |
| MDCK | Madin-Darby Canine Kidney |
| mIMCD3 | Mouse inner medullary collecting duct |
| Na+ | Sodium |
| NCC | Na+ Cl− cotransporter |
| PHA-II | Pseudohypoaldosteronism type II (also known as Gordon’s syndrome) |
| ROMK | Renal outer medullary potassium channel |
| TER | Transepithelial resistance |
| TJ | Tight junction |
| V2R | Vasopressin type 2 receptor |
| WNK4 | With no lysine kinases 4 |
References
- Powell, D.W. Barrier function of epithelia. Am. J. Phys. 1981, 241, G275–G288. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, M.G.; Palade, G.E. Junctional complexes in various epithelia. J. Cell Biol. 1963, 17, 375–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denker, B.M.; Sabath, E. The biology of epithelial cell tight junctions in the kidney. J. Am. Soc. Nephrol. 2011, 22, 622–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef]
- Colegio, O.R.; Van Itallie, C.M.; McCrea, H.J.; Rahner, C.; Anderson, J.M. Claudins create charge-selective channels in the paracellular pathway between epithelial cells. Am. J. Physiol. Cell Physiol. 2002, 283, C142–C147. [Google Scholar] [CrossRef] [Green Version]
- Schneeberger, E.E.; Robert, D.L. The tight junction: A multifunctional complex. Am. J. Physiol. Cell Physiol. 2004, 286, C1213–C1228. [Google Scholar] [CrossRef]
- Diamond, J.M. The epithelial junction: Bridge, gate, and fence. Physiologist 1977, 20, 10–18. [Google Scholar]
- Yu, A.S. Claudins and the kidney. J. Am. Soc. Nephrol. 2015, 26, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Furuse, M.; Sasaki, H.; Tsukita, S. Manner of Interaction of Heterogeneous Claudin Species within and Between Tight Junction Strands. J. Cell Biol. 1999, 147, 891–903. [Google Scholar] [CrossRef]
- Furuse, M.; Sasaki, H.; Fujimoto, K.; Tsukita, S. A Single Gene Product, Claudin-1 or -2, Reconstitutes Tight Junction Strands and Recruits Occludin in Fibroblasts. J. Cell Biol. 1998, 143, 391–401. [Google Scholar] [CrossRef]
- Pearce, D.; Soundararajan, R.; Trimpert, C.; Kashlan, O.B.; Deen, P.M.T.; Kohan, D.E. Collecting duct principal cell transport processes and their regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.; Bhalla, V.; Pastor-Soler, N.M. Intercalated Cells of the Kidney Collecting Duct in Kidney Physiology. Semin. Nephrol. 2019, 39, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Giebisch, G. Regulation of potassium (K) handling in the renal collecting duct. Pflug. Arch. 2009, 458, 157–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, G.; Winkler, L.; Mueller, S.L.; Haseloff, R.F.; Piontek, J.; Blasig, I.E. Structure and function of claudins. Biochim. Biophys. Acta 2008, 1778, 631–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.B.; Lu, Y.; Choate, K.A.; Velazquez, H.; Al-Sabban, E.; Praga, M.; Casari, G.; Bettinelli, A.; Colussi, G.; Rodriguez-Soriano, J.; et al. Paracellin-1, a Renal Tight Junction Protein Required for Paracellular Mg2+Resorption. Science 1999, 285, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Kiuchi-Saishin, Y.; Gotoh, S.; Furuse, M.; Takasuga, A.; Tano, Y.; Tsukita, S. Differential Expression Patterns of Claudins, Tight Junction Membrane Proteins, in Mouse Nephron Segments. J. Am. Soc. Nephrol. 2002, 13, 875–886. [Google Scholar] [PubMed]
- Li, W.Y.; Huey, C.L.; Yu, A.S.L. Expression of claudin-7 and -8 along the mouse nephron. Am. J. Physiol. Ren. Physiol. 2004, 286, F1063–F1071. [Google Scholar] [CrossRef] [Green Version]
- Ziemens, A.; Sonntag, S.R.; Wulfmeyer, V.C.; Edemir, B.; Bleich, M.; Himmerkus, N. Claudin 19 Is Regulated by Extracellular Osmolality in Rat Kidney Inner Medullary Collecting Duct Cells. Int. J. Mol. Sci. 2019, 20, 4401. [Google Scholar] [CrossRef] [Green Version]
- Milatz, S.; Krug, S.M.; Rosenthal, R.; Gunzel, D.; Muller, D.; Schulzke, J.D.; Amasheh, S.; Fromm, M. Claudin-3 acts as a sealing component of the tight junction for ions of either charge and uncharged solutes. Biochim. Biophys. Acta 2010, 1798, 2048–2057. [Google Scholar] [CrossRef] [Green Version]
- Coyne, C.B.; Gambling, T.M.; Boucher, R.C.; Carson, J.L.; Johnson, L.G. Role of claudin interactions in airway tight junctional permeability. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L1166–L1178. [Google Scholar] [CrossRef]
- Schröder, K. Generierung und Charakterisierung ei-nes Claudin-3-defizienten Mausmodells. Humboldt-Universität zu Berlin, Mathematisch-Naturwissenschaftliche Fakultät I. 2013. Available online: https://edoc.hu-berlin.de/handle/18452/17423 (accessed on 23 December 2019).
- Hou, J.; Renigunta, A.; Yang, J.; Waldegger, S. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc. Natl. Acad. Sci. USA 2010, 107, 18010–18015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Yu, M.; Yang, J.; Gonzales, E.; Perez, R.; Hou, M.; Tripathi, P.; Hering-Smith, K.S.; Hamm, L.L.; Hou, J. The Cap1-claudin-4 regulatory pathway is important for renal chloride reabsorption and blood pressure regulation. Proc. Natl. Acad. Sci. USA 2014, 111, E3766–E3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandre, M.D.; Lu, Q.; Chen, Y.H. Overexpression of claudin-7 decreases the paracellular Cl- conductance and increases the paracellular Na+ conductance in LLC-PK1 cells. J. Cell Sci. 2005, 118, 2683–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Gomes, A.S.; Paul, D.L.; Goodenough, D.A. Study of claudin function by RNA interference. J. Biol. Chem. 2006, 281, 36117–36123. [Google Scholar] [CrossRef] [Green Version]
- Tatum, R.; Zhang, Y.; Lu, Q.; Kim, K.; Jeansonne, B.G.; Chen, Y.H. WNK4 phosphorylates ser(206) of claudin-7 and promotes paracellular Cl(-) permeability. FEBS Lett. 2007, 581, 3887–3891. [Google Scholar] [CrossRef] [Green Version]
- Tatum, R.; Zhang, Y.; Salleng, K.; Lu, Z.; Lin, J.J.; Lu, Q.; Jeansonne, B.G.; Ding, L.; Chen, Y.H. Renal salt wasting and chronic dehydration in claudin-7-deficient mice. Am. J. Physiol. Ren. Physiol. 2010, 298, F24–F34. [Google Scholar] [CrossRef] [Green Version]
- Amasheh, S.; Milatz, S.; Krug, S.M.; Bergs, M.; Amasheh, M.; Schulzke, J.D.; Fromm, M. Na+ absorption defends from paracellular back-leakage by claudin-8 upregulation. Biochem. Biophys. Res. Commun. 2009, 378, 45–50. [Google Scholar] [CrossRef]
- Gong, Y.; Wang, J.; Yang, J.; Gonzales, E.; Perez, R.; Hou, J. KLHL3 regulates paracellular chloride transport in the kidney by ubiquitination of claudin-8. Proc. Natl. Acad. Sci. USA 2015, 112, 4340–4345. [Google Scholar] [CrossRef] [Green Version]
- Haddad, N.; El Andalousi, J.; Khairallah, H.; Yu, M.; Ryan, A.K.; Gupta, I.R. The tight junction protein claudin-3 shows conserved expression in the nephric duct and ureteric bud and promotes tubulogenesis in vitro. Am. J. Physiol. Ren. Physiol. 2011, 301, F1057–F1065. [Google Scholar] [CrossRef]
- Fan, J.; Tatum, R.; Hoggard, J.; Chen, Y.H. Claudin-7 Modulates Cl(-) and Na(+) Homeostasis and WNK4 Expression in Renal Collecting Duct Cells. Int. J. Mol. Sci. 2019, 20, 3798. [Google Scholar] [CrossRef] [Green Version]
- Alexandre, M.D.; Jeansonne, B.G.; Renegar, R.H.; Tatum, R.; Chen, Y.H. The first extracellular domain of claudin-7 affects paracellular Cl- permeability. Biochem. Biophys. Res. Commun. 2007, 357, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.; Rahner, C.; Anderson, J.M. Regulated expression of claudin-4 decreases paracellular conductance through a selective decrease in sodium permeability. J. Clin. Investig. 2001, 107, 1319–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Itallie, C.M.; Fanning, A.S.; Anderson, J.M. Reversal of charge selectivity in cation or anion-selective epithelial lines by expression of different claudins. Am. J. Physiol. Ren. Physiol. 2003, 285, F1078–F1084. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.S.; Enck, A.H.; Lencer, W.I.; Schneeberger, E.E. Claudin-8 expression in Madin-Darby canine kidney cells augments the paracellular barrier to cation permeation. J. Biol. Chem. 2003, 278, 17350–17359. [Google Scholar] [CrossRef] [Green Version]
- Konrad, M.; Schaller, A.; Seelow, D.; Pandey, A.V.; Waldegger, S.; Lesslauer, A.; Vitzthum, H.; Suzuki, Y.; Luk, J.M.; Becker, C.; et al. Mutations in the Tight-Junction Gene Claudin 19 (CLDN19) Are Associated with Renal Magnesium Wasting, Renal Failure, and Severe Ocular Involvement. Am. J. Hum. Genet. 2006, 79, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Angelow, S.; El-Husseini, R.; Kanzawa, S.A.; Yu, A.S. Renal localization and function of the tight junction protein, claudin-19. Am. J. Physiol. Ren. Physiol. 2007, 293, F166–F177. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Renigunta, A.; Konrad, M.; Gomes, A.S.; Schneeberger, E.E.; Paul, D.L.; Waldegger, S.; Goodenough, D.A. Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J. Clin. Investig. 2008, 118, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, R.; Milatz, S.; Krug, S.M.; Oelrich, B.; Schulzke, J.D.; Amasheh, S.; Gunzel, D.; Fromm, M. Claudin-2, a component of the tight junction, forms a paracellular water channel. J. Cell Sci. 2010, 123, 1913–1921. [Google Scholar] [CrossRef] [Green Version]
- Knepper, M.A.; Kwon, T.H.; Nielsen, S. Molecular physiology of water balance. N. Engl. J. Med. 2015, 372, 1349–1358. [Google Scholar] [CrossRef]
- Ranieri, M.; Di Mise, A.; Tamma, G.; Valenti, G. Vasopressin-aquaporin-2 pathway: Recent advances in understanding water balance disorders. F1000Res 2019, 8, F1000. [Google Scholar] [CrossRef] [Green Version]
- Loonen, A.J.M.; Knoers, N.V.A.M.; van Os, C.H.; Deen, P.M.T. Aquaporin 2 Mutations in Nephrogenic Diabetes Insipidus. Semin. Nephrol. 2008, 28, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Hinze, C.; Ruffert, J.; Walentin, K.; Himmerkus, N.; Nikpey, E.; Tenstad, O.; Wiig, H.; Mutig, K.; Yurtdas, Z.Y.; Klein, J.D.; et al. GRHL2 Is Required for Collecting Duct Epithelial Barrier Function and Renal Osmoregulation. J. Am. Soc. Nephrol. 2018, 29, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Aue, A.; Hinze, C.; Walentin, K.; Ruffert, J.; Yurtdas, Y.; Werth, M.; Chen, W.; Rabien, A.; Kilic, E.; Schulzke, J.D.; et al. A Grainyhead-Like 2/Ovo-Like 2 Pathway Regulates Renal Epithelial Barrier Function and Lumen Expansion. J. Am. Soc. Nephrol. 2015, 26, 2704–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansom, S.C.; Weinman, E.J.; O’Neil, R.G. Microelectrode assessment of chloride-conductive properties of cortical collecting duct. Am. J. Physiol. Cell Physiol. 1984, 274, F291–F302. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.; Meiselas, L.E.; Auerbach, T. The effect of acute hypovolemia on the release of “aldosterone” and on the renal excretion of sodium. J. Clin. Investig. 1958, 37, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narikiyo, T.; Kitamura, K.; Adachi, M.; Miyoshi, T.; Iwashita, K.; Shiraishi, N.; Nonoguchi, H.; Chen, L.-M.; Chai, K.X.; Chao, J.; et al. Regulation of prostasin by aldosterone in the kidney. J. Clin. Investig. 2002, 109, 401–408. [Google Scholar] [CrossRef]
- Vallet, V.; Chraibi, A.; Gaeggeler, H.-P.; Horisberger, J.-D.; Rossier, B.C. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature 1997, 389, 607–610. [Google Scholar] [CrossRef]
- Yamauchi, K.; Rai, T.K.K.; Sohara, E.; Suzuki, T.; Itoh, T.; Suda, S.; Hayama, A.; Sasaki, S.; Uchida, S. Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc. Natl. Acad. Sci. USA 2004, 101, 4690–4694. [Google Scholar] [CrossRef] [Green Version]
- Kahle, K.T.; MacGregor, G.G.; Wilson, F.H.; Van Hoek, A.N.; Brown, D.; Ardito, T.; Kashgarian, M.; Giebisch, G.; Hebert, S.C.; Boulpaep, E.L.; et al. Paracellular Cl- permeability is regulated by WNK4 kinase: Insight into normal physiology and hypertension. Proc. Natl. Acad. Sci. USA 2004, 101, 14877–14882. [Google Scholar] [CrossRef] [Green Version]
- Le Moellic, C.; Boulkroun, S.; Gonzalez-Nunez, D.; Dublineau, I.; Cluzeaud, F.; Fay, M.; Blot-Chabaud, M.; Farman, N. Aldosterone and tight junctions: Modulation of claudin-4 phosphorylation in renal collecting duct cells. Am. J. Physiol. Cell Physiol. 2005, 289, C1513–C1521. [Google Scholar] [CrossRef] [Green Version]
- Ring, A.M.; Cheng, S.X.; Leng, Q.; Kahle, K.T.; Rinehart, J.; Lalioti, M.D.; Volkman, H.M.; Wilson, F.H.; Hebert, S.C.; Lifton, R.P. WNK4 regulates activity of the epithelial Na+ channel in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 4020–4024. [Google Scholar] [CrossRef] [Green Version]
- Kahle, K.T.; Wilson, F.H.; Leng, Q.; Lalioti, M.D.; O’Connell, A.D.; Dong, K.; Rapson, A.K.; MacGregor, G.G.; Giebisch, G.; Hebert, S.C.; et al. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat. Genet. 2003, 35, 372–376. [Google Scholar] [CrossRef]
- Siragy, H.M.; Awad, A.; Abadir, P.; Webb, R. The angiotensin II type 1 receptor mediates renal interstitial content of tumor necrosis factor-alpha in diabetic rats. Endocrinology 2003, 144, 2229–2233. [Google Scholar] [CrossRef] [Green Version]
- Hollenberg, N.K.; Stevanovic, R.; Agarwal, A.; Lansang, M.C.; Price, D.A.; Laffel, L.M.; Williams, G.H.; Fisher, N.D. Plasma aldosterone concentration in the patient with diabetes mellitus. Kidney Int. 2004, 65, 1435–1439. [Google Scholar] [CrossRef] [Green Version]
- Molina-Jijon, E.; Rodriguez-Munoz, R.; Namorado Mdel, C.; Pedraza-Chaverri, J.; Reyes, J.L. Oxidative stress induces claudin-2 nitration in experimental type 1 diabetic nephropathy. Free Radic. Biol. Med. 2014, 72, 162–175. [Google Scholar] [CrossRef]
- Molina-Jijon, E.; Rodriguez-Munoz, R.; Gonzalez-Ramirez, R.; Namorado-Tonix, C.; Pedraza-Chaverri, J.; Reyes, J.L. Aldosterone signaling regulates the over-expression of claudin-4 and -8 at the distal nephron from type 1 diabetic rats. PLoS ONE 2017, 12, e0177362. [Google Scholar] [CrossRef] [Green Version]
- Canessa, C.M.; Schild, L.; Buell, G.; Thorens, B.; Gautschi, I.; Horisberger, J.-D.; Rossier, B.C. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 1994, 367, 463–467. [Google Scholar] [CrossRef]
- Gordon, R.D. Syndrome of hypertension and hyperkalemia with normal glomerular filtration rate. Hypertension 1986, 8, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Wilson, F.H.; Disse-Nicodeme, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.-M.; et al. Human hypertension caused by mutations in WNK kinase. Science 2001, 293, 1107–1112. [Google Scholar] [CrossRef] [Green Version]
- Boyden, L.M.; Choi, M.; Choate, K.A.; Nelson-Williams, C.J.; Farhi, A.; Toka, H.R.; Tikhonova, I.R.; Bjornson, R.; Mane, S.M.; Colussi, G.; et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012, 482, 98–102. [Google Scholar] [CrossRef]
- Louis-Dit-Picard, H.; Barc, J.; Trujillano, D.; Miserey-Lenkei, S.; Bouatia-Naji, N.; Pylypenko, O.; Beaurain, G.; Bonnefond, A.; Sand, O.; Simian, C.; et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 2012, 44 (Suppl. S1–S3), 456–460. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).






{kind=link}
{kind=link}
{kind=link}