Tight Junctions of the Outer Blood Retina Barrier


Abstract
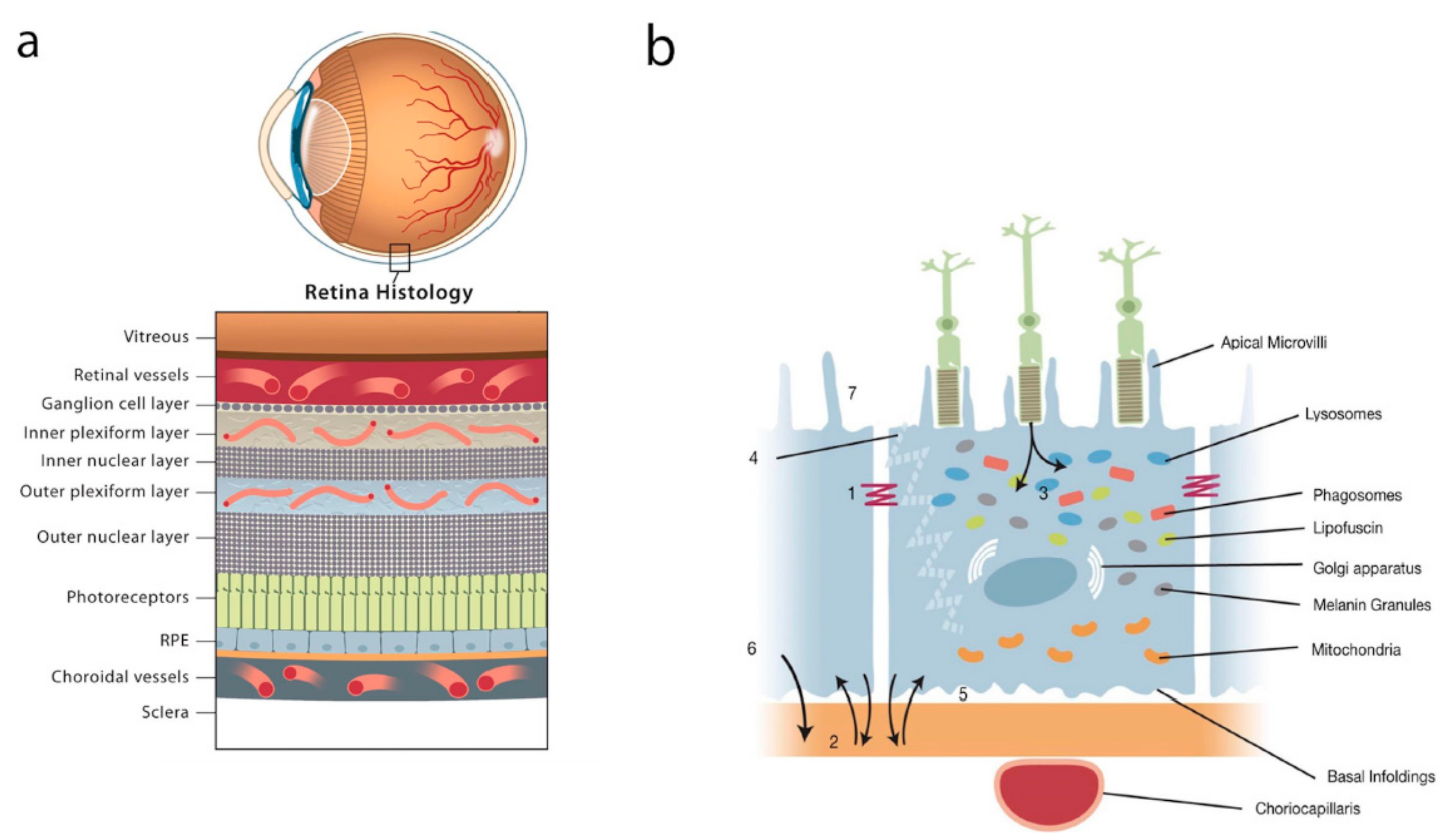
:1. Introduction
2. The Retinal Pigment Epithelium (RPE)
3. Tight Junctions (TJs)
4. Role in Pathology
5. Diabetic Retinopathy
6. Age-Related Macular Degeneration (AMD)
7. Central Serous Chorioretinopathy (CSCR)
8. Sorsby’s Fundus Dystrophy
9. Retinitis Pigmentosa
Mutations in CLDN-19
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cunha-Vaz, J.; Bernardes, R.; Lobo, C. Blood-retinal barrier. Eur. J. Ophthalmol. 2011, 21, 3–9. [Google Scholar] [CrossRef]
- Simó, R.; Villarroel, M.; Corraliza, L.; Hernández, C.; Garcia-Ramírez, M. The Retinal pigment epithelium: Something more than a constituent of the blood-retinal barrier—Implications for the pathogenesis of diabetic retinopathy. J. Biomed. Biotechnol. 2010, 2010, 190724. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Humphries, P. The blood-retina barrier. In Biology and Regulation of Blood-Tissue Barriers; Cheng, C.Y., Ed.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2013; pp. 70–84. ISBN 978-1-4614-4711-5. [Google Scholar]
- Rizzolo, L.J. Development and role of tight junctions in the retinal pigment epithelium. Int. Rev. Cytol. 2007, 258, 195–234. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, A.D.; Finnemann, S.C.; Bonhila, V.L.; Rodriguez-Boulan, E. Morphogenesis of the retinal pigment epithelium: Toward understanding retinal degenerative diseases. Ann. N.Y. Acad. Sci. 1998, 857, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Marneros, A.G.; Fan, J.; Yokoyama, Y.; Gerber, H.P.; Ferrara, N.; Crouch, R.K.; Olsen, B.R. Vascular endothelial growth factor expression in the retinal pigment epithelium Is essential for choriocapillaris development and visual function. Am. J. Pathol. 2005, 167, 1451–1459. [Google Scholar] [CrossRef] [Green Version]
- Bazzoni, G.; Martínez-Estrada, O.M.; Orsenigo, F.; Cordenonsi, M.; Citi, S.; Dejana, E. Interaction of Junctional Adhesion Molecule with the Tight Junction Components ZO-1, Cingulin, and Occludin. J. Biol. Chem. 2000, 275, 20520–20526. [Google Scholar] [CrossRef] [Green Version]
- Bazzoni, G.; Dejana, E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Rao, V.S.; Adelman, R.A.; Rizzolo, L.J. Claudin-19 and the barrier properties of the human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1392–1403. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Adelman, R.A.; Rizzolo, L.J. Minimal effects of VEGF and anti-VEGF drugs on the permeability or selectivity of RPE tight junctions. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3216–3225. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Xiao, W.; Zhu, X.; Mao, Y.; Liu, X.; Chen, X.; Huang, J.; Tang, S.; Rizzollo, L.W. Differential expression of claudins in retinas during normal development and the angiogenesis of oxygen induced retinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7556–7564. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.H.; Fisher, S.K. The relationship of primate foveal cones to the pigment epithelium. J. Ultrastruct. Res. 1979, 67, 23–32. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Hicks, D.; Hamel, C.P. The retinal pigment epithelium in health and disease. Curr. Mol. Med. 2010, 10, 802–823. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Fogg, V.C.; Margolis, B. Tight junctions and cell polarity. Annu. Rev. Cell Dev. Biol. 2006, 22, 207–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [Green Version]
- Besharse, J.C.; Hollyfield, J.G.; Raybron, M.E. Photoreceptor outer segments: Accelerated membrane renewal in rods after exposure to light. Science 1997, 196, 536–538. [Google Scholar] [CrossRef]
- Sakakibara, A.; Furuse, M.; Saitou, M.; Ando-Akatsuka, Y.; Tsukita, S. Possible involvement of phosphorylation of occludin in tight junction formation. J. Cell Biol. 1997, 137, 1393–1401. [Google Scholar] [CrossRef] [Green Version]
- Farquhar, M.G.; Palade, G.E. Junctional complexes in various epithelia. J. Cell Biol. 1963, 17, 375–412. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Coranguez, M.; Liu, X.; Antonetti, D.A. Tight junctions in cell proliferation. Int. J. Mol. Sci. 2019, 20, 5972. [Google Scholar] [CrossRef] [Green Version]
- Raleigh, D.R.; Marchiando, A.M.; Zhang, Y.; Shen, L.; Sasaki, H.; Wang, Y.; Long, M.; Turner, L.R. Tight junction-associated MARVEL proteins marvelD3, tricellulin and occludin have distinct but overlapping functions. Mol. Biol. Cell 2010, 21, 1200–1213. [Google Scholar] [CrossRef] [Green Version]
- Mariano, C.; Sasaki, H.; Brites, D.; Brito, M.A. A look at tricellulin and its role in tight junction formation and maintenance. Eur. J. Cell Biol. 2011, 90, 787–796. [Google Scholar] [CrossRef]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef]
- Furuse, M.; Itoh, T.; Hirase, A.; Nagafuchi, S.; Yonemura, S.; Tsukita, S.; Tsukita, S. Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J. Cell Biol. 1994, 127, 1617–1626. [Google Scholar] [CrossRef] [Green Version]
- Balda, M.S.; Whitney, J.A.; Flores, C.; Gonzalez, S.; Cereijido, M.; Matter, K. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J. Cell Biol. 1996, 134, 1031–1049. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Merzdorf, C.; Paul, D.L.; Goodenough, D.A. COOH terminus of occludin is required for tight junction barrier function in early Xenopus embryos. J. Cell Biol. 1997, 138, 891–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, G.J.; Mullin, J.M.; Ryan, M.P. Occludin: Structure, function and regulation. Adv. Drug Deliv. Rev. 2005, 57, 883–917. [Google Scholar] [CrossRef] [PubMed]
- Saitou, M.; Fujimoto, K.; Doi, Y.; Fujimoto, T.; Furuse, M.; Takano, H.; Noda, T.; Tsukita, S. Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J. Cell Biol. 1998, 141, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Saitou, M.; Furuse, M.; Sasaki, H.; Schulzke, J.D.; Fromm, M.; Takano, H.; Noda, T.; Tsukita, S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell 2000, 11, 4131–4142. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.; Gumbiner, B.M. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J. Cell Biol. 1997, 136, 399–409. [Google Scholar] [CrossRef]
- Rizzolo, L.; Peng, S.; Luo, Y.; Xiao, W. Integration of tight junctions and claudins with the barrier functions of the retinal pigment epithelium. Prog. Retin. Eye Res. 2011, 30, 296–323. [Google Scholar] [CrossRef]
- Traweger, A.; Fang, D.; Liu, Y.C.; Stelzhammer, W.; Krizbai, I.A.; Bauer, H.C.; Bauer, H. The Tight-Junction Specific Protein Occludin is a Functional Target of the E3 Ubiquitin-Protein Ligase Itch. J. Biol. Chem. 2002, 277, 10201–10208. [Google Scholar] [CrossRef] [Green Version]
- Wong, V. Phosphorylation of occludin correlates with occludin localization and function at the tight junction. Am. J. Physiol. 1997, 273, 1859–1867. [Google Scholar] [CrossRef] [PubMed]
- Staddon, J.M.; Smales, C.; Schulze, C.; Esch, F.S.; Rubin, L.L. p120, a p120-related protein (p100), and the cadherin/catenin complex. J. Cell Biol. 1995, 130, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Kale, G.; Naren, A.P.; Sheth, P.; Rao, R.K. Tyrosine phosphorylation of occludin attenuates its interactions with ZO-1, ZO-2, and ZO-3. Biochem. Biophys. Res. Commun. 2003, 302, 324–329. [Google Scholar] [CrossRef]
- Colegio, O.R.; Van Itallie, C.; Rahner, C.; Anderson, J.M. Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fibril architecture. Am. J. Physiol. Cell Physiol. 2003, 284, 1346–1354. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Van Itallie, C.; McCrea, H.J. Claudins create charge-selective channels in the paracellular pathway between epithelial cells. Am. J. Physiol. Cell Physiol. 2002, 282, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Van Italie, C.M.; Anderson, J.M. Claudins and paracellular transport. Annu. Rev. Physiol. 2006, 68, 403–429. [Google Scholar] [CrossRef]
- Morita, K.; Sasaki, H.; Furuse, M.; Tsukita, S. Endothelial claudin: Claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 1999, 147, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Furuse, M.; Morita, K.; Kubota, K.; Saitou, M.; Tsukita, S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J. Cell Biol. 1999, 147, 1351–1363. [Google Scholar] [CrossRef] [Green Version]
- Konrad, M.; Schaller, A.; Seelow, D.; Pandey, A.V.; Waldegger, S.; Lesslauer, A.; Vitzthum, H.; Suzuki, Y.; Luk, J.M.; Becker, C.; et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am. J. Hum. Genet. 2006, 79, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.B.; Xu, T.; Peng, S.; Singh, D.; Ghiassi-Nejed, M.; Adelman, R.A.; Rizzolo, L.J. Disease-associated mutations of claudin-19 disrupt retinal neurogenesis and visual function. Commun. Biol. 2019, 2, 113. [Google Scholar] [CrossRef]
- Mandell, K.J.; Parkos, C.A. The JAM family of proteins. Adv. Drug Deliv. Rev. 2005, 57, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Mariscal, L.; Betanzos, A.; Avila-Flores, A. MAGUK proteins: Structure and role in the tight junction. Semin. Cell. Dev. Biol. 2000, 11, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, B.R.; Siliciano, J.D.; Mooseker, M.S.; Goodenough, D.A. Identification of ZO-1: A high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J. Cell Biol. 1986, 103, 755–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef]
- Muller, S.L.; Portwich, M.; Schmidt, A.; Utepbergenov, D.I.; Huber, O.; Blasig, I.E.; Krause, G. The tight junction protein occludin and the adherens junction protein α-catenin share a common interaction mechanism with ZO-1. J. Biol. Chem. 2005, 280, 3747–3756. [Google Scholar] [CrossRef] [Green Version]
- Balda, M.S.; Matter, K. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO. J. 2000, 19, 2024–2033. [Google Scholar] [CrossRef]
- Balda, M.S.; Garrett, M.D.; Matter, K. The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J. Cell Biol. 2003, 160, 423–432. [Google Scholar] [CrossRef]
- Vinores, S.A.; Gadegbeku, C.; Campochiaro, P.A.; Green, W.R. Immunohistochemical localization of blood-retinal barrier breakdown in human diabetics. Am. J. Pathol. 1989, 134, 231–235. [Google Scholar] [CrossRef]
- Xia, T.; Rizzolo, L.J. Effects of diabetic retinopathy on the barrier functions of the retinal pigment epothelium. Vis. Res. 2017, 129, 72–81. [Google Scholar] [CrossRef]
- Xu, H.Z.; Le, Y.Z. Significant of outer blood-retina breakdown in diabetes and ischaemia. Investig. Ophthalmol. Vis. Sci. 2011, 53, 2160–2164. [Google Scholar] [CrossRef]
- Desjardins, D.M.; Yates, P.W.; Dahrouj, M.; Yiu, Y.; Crosson, C.E.; Ablonczy, Z. Progressive early breakdown of retinal pigment epithelium function in hyperglycaemic rats. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2706–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, U.; Evans, J.; Rosenfeld, P.J. Age related macular degeneration. BMJ 2010, 340, 981. [Google Scholar] [CrossRef] [PubMed]
- Cook, H.L.; Pater, P.J.; Tufail, A. Age-related macular degeneration: Diagnosis and management. Br. Med. Bull. 2008, 85, 127–149. [Google Scholar] [CrossRef]
- Fleckenstein, M.; Mitchell, P.; Freund, K.B.; Sadda, S.; Holz, F.G.; Brittain, C.; Henry, E.C.; Ferrara, D. The progression of geographic atrophy secondary to age-related macular degeneration. Ophthalmology 2018, 125, 369–390. [Google Scholar] [CrossRef]
- Ramrattan, R.S.; van der Schaft, T.L.; Mooy, C.M.; de Bruijn, W.C.; Mulder, P.G.; de Jong, P.T. Morphometric analysis of Bruch’s membrane, the choriocapillaris, and the choroid in aging. Investig. Ophthalmol. Vis. Sci. 1994, 35, 2857–2864. [Google Scholar]
- Chen, M.; Xu, H. Parainflammation, chronic inflammation, and age related macular degeneration. J. Leukoc. Biol. 2015, 98, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Brassler, S.B. Introduction: Understanding the role of angiogenesis and antiangiogenic agents in age-related macular degeneration. Ophthalmology 2009, 116, S1–S7. [Google Scholar] [CrossRef]
- Farjood, F.; Vargis, E. Physical disruption of cell-cell contact induced VEGF expression in RPE cells. Mol. Vis. 2017, 23, 431–446, pmc:5524271. [Google Scholar]
- Wang, M.; Munch, I.C.; Hasler, P.W.; Prunte, C.; Larsen, M. Central serous retinopathy. Acta. Ophthalmol. Scand. 2008, 86, 126–145. [Google Scholar] [CrossRef]
- Nicholson, B.; Noble, J.; Forooghian, F.; Meyerle, C. Central serous chorioretinopathy: Update on pathophysiology and treatment. Surv. Ophthalmol. 2014, 58, 103–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, D.R.G.; Brown, F.E.; Cree, A.J.; Ratnayaka, J.A.; Lotery, A.J. Sorsby fundus dystrophy—A review of pathology and disease mechanisms. Exp. Eye Res. 2017, 165, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis Pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Hamel, C. Retinitis Pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef]
- Campbell, M.; Humphries, M.; Kennan, A.; Kenna, P.; Humphries, P.; Brankin, B. Aberrant retinal tight junction and adherens junction protein expression in an animal model of autosomal recessive Retinitis pigmentosa: The Rho (−/−) mouse. Exp. Eye Res. 2006, 83, 484–492. [Google Scholar] [CrossRef]
- Falasconi, F.; Biagioni, M.; Novelli, E.; Piabo, I.; Gargini, C.; Strettoi, E. Retinal phenotype in the rd9 mutant mouse, a model of X-linked RP. Front. Neurosci. 2019, 13, 991. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Kini, A.; Wang, Y.; Liu, T.; Chen, Y.; Vukmanic, E.; Emery, D.; Liu, Y.; Jin, L.; Lee, S.J.; et al. Metabolic deregulation of the blood-outer retinal barrier in retinitis pigmentosa. Cell Rep. 2019, 28, 1323–1334. [Google Scholar] [CrossRef] [Green Version]
- Vinores, S.A.; Kuchle, M.; Derevjanik, N.L.; Henderer, J.D.; Mahlow, J.; Green, W.R.; Campochiaro, P.A. Blood-retinal barrier breakdown in retinitis pigmentosa: Light and electron microscopic immunolocalization. Histol. Histopathol. 1995, 10, 913–923. [Google Scholar]
- Strong, S.; Liew, G.; Michaelides, M. Retinitis pigmentosa-associated cystoid macular oedema: Pathogenesis and avenues of intervention. Br. J. Ophthalmol. 2017, 101, 31–37. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Outer Blood Retinal Barrier | Inner Blood Retinal Barrier |
|---|---|
| Formed by tight junctions (TJ) between neighbouring retinal pigment epithelium (RPE) cells [1]. Rests on underlying Bruch’s membrane [1] | Formed by TJ between neighbouring retinal endothelial cells [1]. Rests on a basal lamina that is covered by the processes of astrocytes and Müller cells [1] |
| Regulates the paracellular movement of fluids and molecules between the choriocapillaris and the retina [1] | Regulates the paracellular movement of fluids and molecules across retinal capillaries [1] |
| Claudin-19 is the predominant claudin [9], claudin-3 and -10 are also expressed [10] | Claudin-5 is the most predominant claudin, claudin-1 and -2 are also expressed [11] |
| Plays a fundamental role in the microenvironment of the outer retina [1] including regulating access of nutrients from blood to photoreceptors (PRs), eliminating waste products, and maintaining retinal adhesion [1] | Plays a fundamental role in the microenvironment of the neural retina [1] |
| The relationship between the RPE apical villi and PR is considered to be crucial in maintaining visual function [1] | Regulatory signals of the retinal neuronal circuitry are transmitted by astrocytes, muller cells and pericytes thereby influencing the activity of the iBRB [1] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naylor, A.; Hopkins, A.; Hudson, N.; Campbell, M. Tight Junctions of the Outer Blood Retina Barrier. Int. J. Mol. Sci. 2020, 21, 211. https://doi.org/10.3390/ijms21010211
Naylor A, Hopkins A, Hudson N, Campbell M. Tight Junctions of the Outer Blood Retina Barrier. International Journal of Molecular Sciences. 2020; 21(1):211. https://doi.org/10.3390/ijms21010211
Chicago/Turabian StyleNaylor, Aisling, Alan Hopkins, Natalie Hudson, and Matthew Campbell. 2020. "Tight Junctions of the Outer Blood Retina Barrier" International Journal of Molecular Sciences 21, no. 1: 211. https://doi.org/10.3390/ijms21010211
APA StyleNaylor, A., Hopkins, A., Hudson, N., & Campbell, M. (2020). Tight Junctions of the Outer Blood Retina Barrier. International Journal of Molecular Sciences, 21(1), 211. https://doi.org/10.3390/ijms21010211