Type 2 Diabetes Alters Intracellular Ca2+ Handling in Native Endothelium of Excised Rat Aorta

 , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. Somatic and Biochemical Characteristics of OZDF Rats
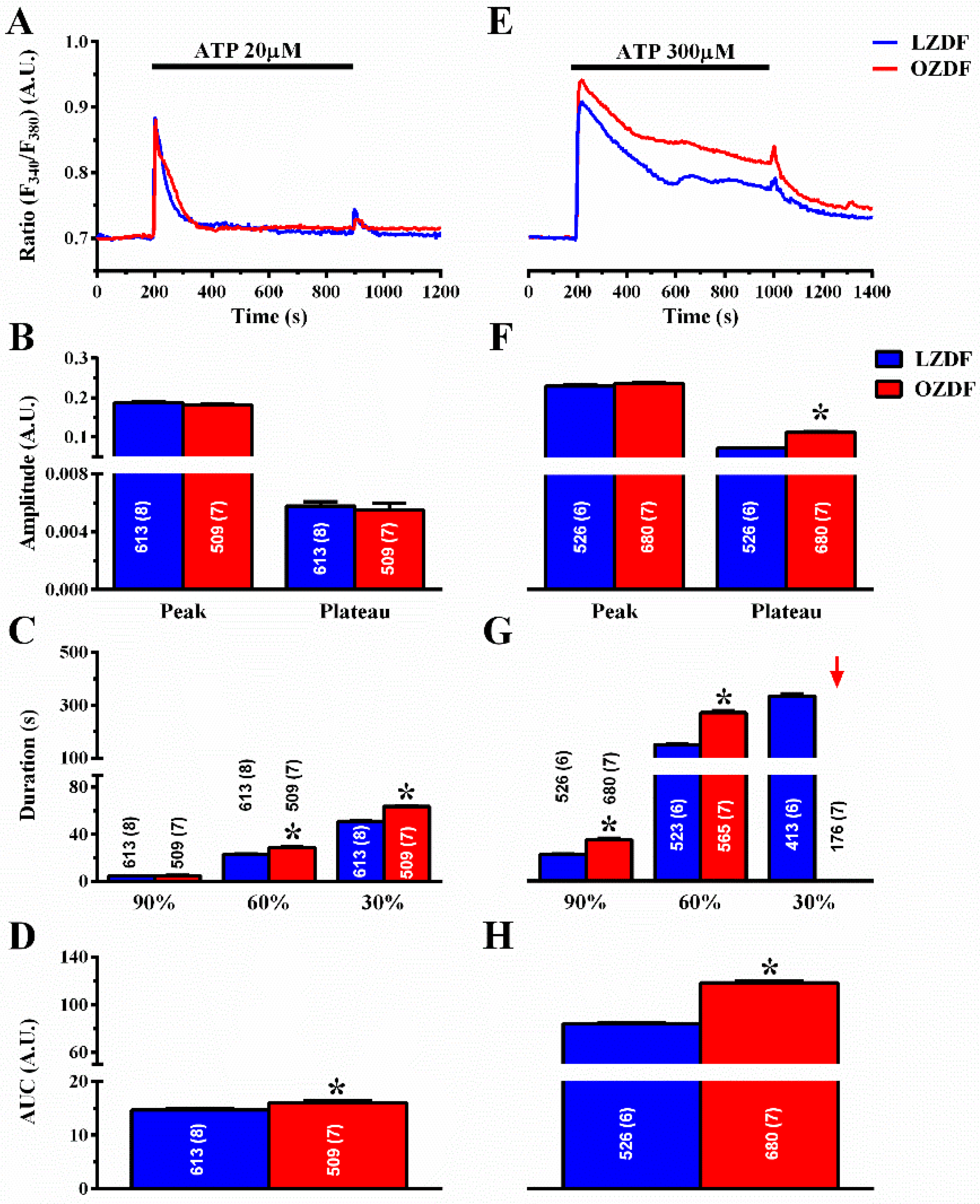
2.2. The Ca2+ Response to Adenosine Triphosphate (ATP) is Enhanced in Native Aortic Endothelium of Diabetic Rats
2.3. The Rate of Decay of Intracellular Ca2+ Release Is Shortened and SOCE Amplitude Is Reduced in Native Aortic Endothelium of OZDF Rats
2.4. The Down-Regulation of SERCA Activity Enhances Plateau Amplitude and Prolongs the Duration of the Ca2+ Response to High Doses of ATP in Native Aortic Endothelium of OZDF Rats
2.5. PMCA Activity Is Not Altered in Native Aortic Endothelium of OZDF Rats
2.6. NCX Activity Is Not Altered in Native Aortic Endothelium of OZDF Rats
2.7. SERCA2B Protein Is Up-Regulated in in Native Aortic Endothelium of OZDF Rats
2.8. ROS Inhibition Rescues SERCA2B-Dependent Ca2+ Sequestration in Native Aortic Endothelium of OZDF Rats
2.9. Evidence for the Contribution of K+ Channels in the Alteration of Intracellular Ca2+ Dynamics in Native Aortic Endothelium of OZDF Rats
3. Discussion
3.1. Ca2+ Clearing in LZDF Rats: the Control Condition
3.2. Evidence that SERCA2B Activity Is Slowed Down in Native Aortic Endothelium of LZDF Rats
3.3. Preliminary Evidence that Ca2+-Dependent K+ Channels Contribute to the Enhanced Ca2+ Response to ATP
3.4. How the Impairment of SERCA2B Activity Could Result in Endothelial Dysfunction in Type 2 Diabetes Mellitus
4. Materials and Methods
4.1. Animals
4.2. Morphometric Parameters
4.3. Oral Glucose Tolerance Test and Insulin Response, Insulin Tolerance Test and Insulin Resistance
4.4. Dissection of the Aorta, Blood and Epididymal Fat Samples
4.5. Solutions
4.6. Intracellular Ca2+ Concentration Measurements
4.7. Data Analysis
4.8. Chemicals
4.9. Immunohistochemistry
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| A.U. | Arbitrary units |
| AUC | Area under de curve |
| ATP | Adenosine triphosphate |
| BMI | Body mass index |
| HOMA-IR | Homeostasis model assessment insulin resistance |
| ITT | Insulin tolerance test |
| [Ca2+]i | Intracellular Ca2+ concentration |
| CE | Carboxyeosin |
| CPA | Cyclopiazonic acid |
| CV | Cardiovascular |
| ER | Endoplasmic reticulum |
| HDL-C | High-density lipoprotein cholesterol |
| InsP3 | Inositol-1,4,5-trisphosphate |
| KBR | KBR-79433 |
| LDL-C | Low-density lipoprotein cholesterol |
| LZDF | Lean Zucker Diabetic Fatty rats |
| NCX | Na+/Ca2+ exchanger |
| OGTT | Oral glucose tolerance test |
| OZDF | Obese Zucker Diabetic Fatty rats |
| PMCA | Plasma membrane Ca2+-ATPase |
| PSS | Physiological salt solution |
| SEA | SEA0400 |
| SERCA | Sarco-endoplasmic reticulum Ca2+-ATPase |
| SOCE | Store-operated Ca2+ entry |
| TG | Thapsigargin |
| VLDL | Very low-density lipoprotein |
References
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.C.; Porter, K.E. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diabetes Vasc. Dis. Res. 2013, 10, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Vascular complications in diabetes mellitus: The role of endothelial dysfunction. Clin. Sci. 2005, 109, 143–159. [Google Scholar] [CrossRef]
- Winer, N.; Sowers, J.R. Epidemiology of Diabetes. J. Clin. Pharmacol. 2004, 44, 397–405. [Google Scholar] [CrossRef]
- Fowler, M.J. Microvascular and Macrovascular Complications of Diabetes. Clin. Diabetes 2011, 29, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Tabit, C.E.; Chung, W.B.; Hamburg, N.M.; Vita, J.A. Endothelial dysfunction in diabetes mellitus: Molecular mechanisms and clinical implications. Rev. Endocr. Metab. Disord. 2010, 11, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Cheng, R.; Ma, J. Angiogenesis in diabetes and obesity. Rev. Endocr. Metab. Disord. 2015, 16, 67–75. [Google Scholar] [CrossRef] [Green Version]
- McCarron, J.G.; Lee, M.D.; Wilson, C. The Endothelium Solves Problems That Endothelial Cells Do Not Know Exist. Trends Pharmacol. Sci. 2017, 38, 322–338. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F. Update on vascular endothelial Ca2+ signalling: A tale of ion channels, pumps and transporters. World J. Biol. Chem. 2012, 3, 127. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Tanzi, F.; Munaron, L. Endothelial Remodelling and Intracellular Calcium Machinery. Curr. Mol. Med. 2014, 14, 457–480. [Google Scholar] [CrossRef] [PubMed]
- Altaany, Z.; Moccia, F.; Munaron, L.; Mancardi, D.; Wang, R. Hydrogen Sulfide and Endothelial Dysfunction: Relationship with Nitric Oxide. Curr. Med. Chem. 2014, 21, 3646–3661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef] [Green Version]
- Khaddaj Mallat, R.; Mathew John, C.; Kendrick, D.J.; Braun, A.P. The vascular endothelium: A regulator of arterial tone and interface for the immune system. Crit. Rev. Clin. Lab. Sci. 2017, 54, 458–470. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a Physiologic Vasorelaxant: Hypertension in Mice with Deletion of Cystathionine-Lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Vanhoutte, P.M.; Tang, E.H.C. Endothelium-dependent contractions: When a good guy turns bad! J. Physiol. 2008, 586, 5295–5304. [Google Scholar] [CrossRef]
- Yakubu, M.A.; Leffler, C.W. L-type voltage-dependent Ca2+ channels in cerebral microvascular endothelial cells and ET-1 biosynthesis. Am. J. Physiol. Cell Physiol. 2002, 283, C1687–C1695. [Google Scholar] [CrossRef] [Green Version]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 Mediate CRAC Currents and Store-Operated Calcium Entry Important for Endothelial Cell Proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef]
- Sánchez-Hernández, Y.; Laforenza, U.; Bonetti, E.; Fontana, J.; Dragoni, S.; Russo, M.; Avelino-Cruz, J.E.; Schinelli, S.; Testa, D.; Guerra, G.; et al. Store-Operated Ca2+ Entry Is Expressed in Human Endothelial Progenitor Cells. Stem Cells Dev. 2010, 19, 1967–1981. [Google Scholar] [CrossRef] [Green Version]
- Berra-Romani, R.; Raqeeb, A.; Guzman-Silva, A.; Torres-Jácome, J.; Tanzi, F.; Moccia, F. Na+–Ca2+ exchanger contributes to Ca2+ extrusion in ATP-stimulated endothelium of intact rat aorta. Biochem. Biophys. Res. Commun. 2010, 395, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Dömötör, E.; Abbott, N.J.; Adam-Vizi, V. Na+–Ca2+ exchange and its implications for calcium homeostasis in primary cultured rat brain microvascular endothelial cells. J. Physiol. 1999, 515, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Baruffi, S.; Spaggiari, S.; Signorelli, S.; CASTELLI, L.; Magistretti, J.; Taglietti, V.; Tanzi, F. Ca2+ uptake by the endoplasmic reticulum Ca2+-ATPase in rat microvascular endothelial cells. Biochem. J. 2002, 364, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Reznick, S.; Li, P.; Liang, W.; van Breemen, C. Ca2+ removal mechanisms in freshly isolated rabbit aortic endothelial cells. Cell Calcium 2002, 31, 265–277. [Google Scholar] [CrossRef]
- Chalmers, S.; McCarron, J.G. The mitochondrial membrane potential and Ca2+ oscillations in smooth muscle. J. Cell Sci. 2008, 121, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Hajnóczky, G.; Hager, R.; Thomas, A.P. Mitochondria Suppress Local Feedback Activation of Inositol 1,4,5-Trisphosphate Receptors by Ca2+. J. Biol. Chem. 1999, 274, 14157–14162. [Google Scholar] [CrossRef] [Green Version]
- Lodola, F.; Laforenza, U.; Cattaneo, F.; Ruffinatti, F.A.; Poletto, V.; Massa, M.; Tancredi, R.; Zuccolo, E.; Khdar, D.A.; Riccardi, A.; et al. VEGF-induced intracellular Ca2+ oscillations are down-regulated and do not stimulate angiogenesis in breast cancer-derived endothelial colony forming cells. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F. Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef] [Green Version]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Reforgiato, M.; Poletto, V.; Lodola, F.; Bottino, C.; Guido, D.; Rappa, A.; Pareek, S.; et al. Enhanced Expression of Stim, Orai, and TRPC Transcripts and Proteins in Endothelial Progenitor Cells Isolated from Patients with Primary Myelofibrosis. PLoS ONE 2014, 9, e91099. [Google Scholar] [CrossRef]
- Fonseca, A.C.R.G.; Moreira, P.I.; Oliveira, C.R.; Cardoso, S.M.; Pinton, P.; Pereira, C.F. Amyloid-Beta Disrupts Calcium and Redox Homeostasis in Brain Endothelial Cells. Mol. Neurobiol. 2015, 51, 610–622. [Google Scholar] [CrossRef]
- Prendergast, C.; Quayle, J.; Burdyga, T.; Wray, S. Atherosclerosis affects calcium signalling in endothelial cells from apolipoprotein E knockout mice before plaque formation. Cell Calcium 2014, 55, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Perrier, E.; Fournet-Bourguignon, M.-P.; Royere, E.; Molez, S.; Reure, H.; Lesage, L.; Gosgnach, W.; Frapart, Y.; Boucher, J.-L.; Villeneuve, N.; et al. Effect of uncoupling endothelial nitric oxide synthase on calcium homeostasis in aged porcine endothelial cells. Cardiovasc. Res. 2009, 82, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Fasanaro, P.; Magenta, A.; Zaccagnini, G.; Cicchillitti, L.; Fucile, S.; Eusebi, F.; Biglioli, P.; Capogrossi, M.C.; Martelli, F. Cyclin D1 degradation enhances endothelial cell survival upon oxidative stress. FASEB J. 2006, 20, 1242–1244. [Google Scholar] [CrossRef] [PubMed]
- Gandhirajan, R.K.; Meng, S.; Chandramoorthy, H.C.; Mallilankaraman, K.; Mancarella, S.; Gao, H.; Razmpour, R.; Yang, X.-F.; Houser, S.R.; Chen, J.; et al. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J. Clin. Investig. 2013. [Google Scholar] [CrossRef]
- Al-awar, A.; Kupai, K.; Veszelka, M.; Szűcs, G.; Attieh, Z.; Murlasits, Z.; Török, S.; Pósa, A.; Varga, C. Experimental Diabetes Mellitus in Different Animal Models. J. Diabetes Res. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Berra-Romani, R.; Raqeeb, A.; Avelino-Cruz, J.E.; Moccia, F.; Oldani, A.; Speroni, F.; Taglietti, V.; Tanzi, F. Ca2+ signaling in injured in situ endothelium of rat aorta. Cell Calcium 2008, 44, 298–309. [Google Scholar] [CrossRef]
- Bondarenko, A. Sodium-calcium exchanger contributes to membrane hyperpolarization of intact endothelial cells from rat aorta during acetylcholine stimulation. Br. J. Pharmacol. 2004, 143, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Usachev, Y.M.; Marchenko, S.M.; Sage, S.O. Cytosolic calcium concentration in resting and stimulated endothelium of excised intact rat aorta. J. Physiol. 1995, 489, 309–317. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Rinaldi, C.; Raqeeb, A.; Castelli, L.; Magistretti, J.; Taglietti, V.; Tanzi, F. The Duration and Amplitude of the Plateau Phase of ATP- and ADP-Evoked Ca2+ Signals Are Modulated by Ectonucleotidases in in situ Endothelial Cells of Rat Aorta. J. Vasc. Res. 2004, 41, 166–173. [Google Scholar] [CrossRef]
- Moccia, F.; Bertoni, G.; Florio Pla, A.; Dragoni, S.; Pupo, E.; Merlino, A.; Mancardi, D.; Munaron, L.; Tanzi, F. Hydrogen Sulfide Regulates Intracellular Ca2+ Concentration in Endothelial Cells from Excised Rat Aorta. Curr. Pharm. Biotechnol. 2011, 12, 1416–1426. [Google Scholar] [CrossRef]
- Lipskaia, L.; Chemaly, E.R.; Hadri, L.; Lompre, A.-M.; Hajjar, R.J. Sarcoplasmic reticulum Ca(2+) ATPase as a therapeutic target for heart failure. Expert Opin. Biol. Ther. 2010, 10, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwathmey, J.K.; Copelas, L.; MacKinnon, R.; Schoen, F.J.; Feldman, M.D.; Grossman, W.; Morgan, J.P. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ. Res. 1987, 61, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccolo, E.; Lim, D.; Kheder, D.A.; Perna, A.; Catarsi, P.; Botta, L.; Rosti, V.; Riboni, L.; Sancini, G.; Tanzi, F.; et al. Acetylcholine induces intracellular Ca2+ oscillations and nitric oxide release in mouse brain endothelial cells. Cell Calcium 2017, 66, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Pierro, C.; Cook, S.J.; Foets, T.C.F.; Bootman, M.D.; Roderick, H.L. Oncogenic K-Ras suppresses IP3-dependent Ca2+ release through remodelling of the isoform composition of IP3Rs and ER luminal Ca2+ levels in colorectal cancer cell lines. J. Cell Sci. 2014, 127, 1607–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lillo, M.A.; Gaete, P.S.; Puebla, M.; Ardiles, N.M.; Poblete, I.; Becerra, A.; Simon, F.; Figueroa, X.F. Critical contribution of Na+–Ca2+ exchanger to the Ca2+-mediated vasodilation activated in endothelial cells of resistance arteries. FASEB J. 2018, 32, 2137–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Endothelial Ca2+ Signaling, Angiogenesis and Vasculogenesis: Just What It Takes to Make a Blood Vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef] [Green Version]
- Horáková, L.; Strosova, M.K.; Spickett, C.M.; Blaskovic, D. Impairment of calcium ATPases by high glucose and potential pharmacological protection. Free Radic. Res. 2013, 47, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Hou, X.; Jourd’heuil, D.; Weisbrod, R.M.; Cohen, R.A. Upregulation of Nox4 by TGFβ1 Oxidizes SERCA and Inhibits NO in Arterial Smooth Muscle of the Prediabetic Zucker Rat. Circ. Res. 2010, 107, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Chinen, I.; Shimabukuro, M.; Yamakawa, K.; Higa, N.; Matsuzaki, T.; Noguchi, K.; Ueda, S.; Sakanashi, M.; Takasu, N. Vascular Lipotoxicity: Endothelial Dysfunction via Fatty-Acid-Induced Reactive Oxygen Species Overproduction in Obese Zucker Diabetic Fatty Rats. Endocrinology 2007, 148, 160–165. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Berra-Romani, R. Targeting the endothelial Ca2+ tool kit to rescue endothelial dysfunction in obesity associated-hypertension. Curr. Med. Chem. 2019, 26. [Google Scholar] [CrossRef]
- Bishara, N.B.; Ding, H. Glucose enhances expression of TRPC1 and calcium entry in endothelial cells. Am. J. Physiol. Circ. Physiol. 2010, 298, H171–H178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daskoulidou, N.; Zeng, B.; Berglund, L.M.; Jiang, H.; Chen, G.-L.; Kotova, O.; Bhandari, S.; Ayoola, J.; Griffin, S.; Atkin, S.L.; et al. High glucose enhances store-operated calcium entry by upregulating ORAI/STIM via calcineurin-NFAT signalling. J. Mol. Med. 2015, 93, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.L.; Taylor, H.; et al. Orai1 and CRAC Channel Dependence of VEGF-Activated Ca 2+ Entry and Endothelial Tube Formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, A.Q.; Hurley, J.R.; Huang, W.; Taghian, T.; Kogan, A.; Cho, H.; Wang, Y.; Narmoneva, D.A. Diabetes Alters Intracellular Calcium Transients in Cardiac Endothelial Cells. PLoS ONE 2012, 7, e36840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giuro, C.M.L.; Shrestha, N.; Malli, R.; Groschner, K.; van Breemen, C.; Fameli, N. Na+/Ca2+ exchangers and Orai channels jointly refill endoplasmic reticulum (ER) Ca2+ via ER nanojunctions in vascular endothelial cells. Pflügers Arch. Eur. J. Physiol. 2017, 469, 1287–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jousset, H.; Frieden, M.; Demaurex, N. STIM1 Knockdown Reveals That Store-operated Ca2+ Channels Located Close to Sarco/Endoplasmic Ca2+ ATPases (SERCA) Pumps Silently Refill the Endoplasmic Reticulum. J. Biol. Chem. 2007, 282, 11456–11464. [Google Scholar] [CrossRef] [Green Version]
- Manjarrés, I.M.; Alonso, M.T.; García-Sancho, J. Calcium entry-calcium refilling (CECR) coupling between store-operated Ca2+ entry and sarco/endoplasmic reticulum Ca2+-ATPase. Cell Calcium 2011, 49, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Manjarrés, I.M.; Rodríguez-García, A.; Alonso, M.T.; García-Sancho, J. The sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) is the third element in capacitative calcium entry. Cell Calcium 2010, 47, 412–418. [Google Scholar] [CrossRef] [Green Version]
- Klishin, A.; Sedova, M.; Blatter, L.A. Time-dependent modulation of capacitative Ca2+ entry signals by plasma membrane Ca2+ pump in endothelium. Am. J. Physiol. Cell Physiol. 1998, 274, C1117–C1128. [Google Scholar] [CrossRef]
- Snitsarev, V.A.; Taylor, C.W. Overshooting cytosolic Ca2+ signals evoked by capacitative Ca2+ entry result from delayed stimulation of a plasma membrane Ca2+ pump. Cell Calcium 1999, 25, 409–417. [Google Scholar] [CrossRef]
- Hughes, E.; Lee, A.K.; Tse, A. Dominant Role of Sarcoendoplasmic Reticulum Ca2+-ATPase Pump in Ca2+ Homeostasis and Exocytosis in Rat Pancreatic β-Cells. Endocrinology 2006, 147, 1396–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredersdorf, S.; Thumann, C.; Zimmermann, W.H.; Vetter, R.; Graf, T.; Luchner, A.; Riegger, G.A.; Schunkert, H.; Eschenhagen, T.; Weil, J. Increased myocardial SERCA expression in early type 2 diabetes mellitus is insulin dependent: In vivo and in vitro data. Cardiovasc. Diabetol. 2012, 11, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwabara, A.; Satoh, M.; Tomita, N.; Sasaki, T.; Kashihara, N. Deterioration of glomerular endothelial surface layer induced by oxidative stress is implicated in altered permeability of macromolecules in Zucker fatty rats. Diabetologia 2010, 53, 2056–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschöpe, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.M.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure with Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev; Talanov; Starinets; Agafonov; Dubinin; Belosludtseva Transport of Ca2+ and Ca2+-Dependent Permeability Transition in Rat Liver Mitochondria under the Streptozotocin-Induced Type I Diabetes. Cells 2019, 8, 1014. [CrossRef] [PubMed] [Green Version]
- Suarez, J.; Cividini, F.; Scott, B.T.; Lehmann, K.; Diaz-Juarez, J.; Diemer, T.; Dai, A.; Suarez, J.A.; Jain, M.; Dillmann, W.H. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J. Biol. Chem. 2018, 293, 8182–8195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.; Lee, M.D.; Heathcote, H.R.; Zhang, X.; Buckley, C.; Girkin, J.M.; Saunter, C.D.; McCarron, J.G. Mitochondrial ATP production provides long-range control of endothelial inositol trisphosphate-evoked calcium signaling. J. Biol. Chem. 2019, 294, 737–758. [Google Scholar] [CrossRef] [Green Version]
- Malli, R.; Frieden, M.; Osibow, K.; Zoratti, C.; Mayer, M.; Demaurex, N.; Graier, W.F. Sustained Ca2+ transfer across mitochondria is Essential for mitochondrial Ca2+ buffering, sore-operated Ca2+ entry, and Ca2+ store refilling. J. Biol. Chem. 2003, 278, 44769–44779. [Google Scholar] [CrossRef] [Green Version]
- Malli, R.; Frieden, M.; Trenker, M.; Graier, W.F. The role of mitochondria for Ca2+ refilling of the endoplasmic reticulum. J. Biol. Chem. 2005, 280, 12114–12122. [Google Scholar] [CrossRef] [Green Version]
- Climent, B.; Moreno, L.; Martínez, P.; Contreras, C.; Sánchez, A.; Pérez-Vizcaíno, F.; García-Sacristán, A.; Rivera, L.; Prieto, D. Upregulation of SK3 and IK1 Channels Contributes to the Enhanced Endothelial Calcium Signaling and the Preserved Coronary Relaxation in Obese Zucker Rats. PLoS ONE 2014, 9, e109432. [Google Scholar] [CrossRef] [Green Version]
- Schach, C.; Resch, M.; Schmid, P.M.; Riegger, G.A.; Endemann, D.H. Type 2 diabetes: Increased expression and contribution of IK Ca channels to vasodilation in small mesenteric arteries of ZDF rats. Am. J. Physiol. Circ. Physiol. 2014, 307, H1093–H1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilius, B.; Droogmans, G. Ion channels and their functional role in vascular endothelium. Physiol. Rev. 2001, 81, 1415–1459. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, Y.; Han, S.-Z.; Kim, S.; Akishita, M.; Kozaki, K.; Toba, K.; Orimo, H. Augmented Contractile Function and Abnormal Ca2+ Handling in the Aorta of Zucker Obese Rats with Insulin Resistance. Diabetes 1996, 45, S55–S58. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.; Contreras, C.; Climent, B.; Gutiérrez, A.; Muñoz, M.; García-Sacristán, A.; López, M.; Rivera, L.; Prieto, D. Impaired Ca2+ handling in resistance arteries from genetically obese Zucker rats: Role of the PI3K, ERK1/2 and PKC signaling pathways. Biochem. Pharmacol. 2018, 152, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Villalba, N.; Contreras, C.; Hernández, M.; García-Sacristán, A.; Prieto, D. Impaired Ca2+ handling in penile arteries from prediabetic Zucker rats: Involvement of Rho kinase. Am. J. Physiol. Circ. Physiol. 2011, 300, H2044–H2053. [Google Scholar] [CrossRef]
- Howarth, F.C.; Qureshi, M.A.; Hassan, Z.; Isaev, D.; Parekh, K.; John, A.; Oz, M.; Raza, H.; Adeghate, E.; Adrian, T.E. Contractility of ventricular myocytes is well preserved despite altered mechanisms of Ca2+ transport and a changing pattern of mRNA in aged type 2 Zucker diabetic fatty rat heart. Mol. Cell. Biochem. 2012, 361, 267–280. [Google Scholar] [CrossRef]
- Bréchard, S.; Tschirhart, E.J. Regulation of superoxide production in neutrophils: Role of calcium influx. J. Leukoc. Biol. 2008, 84, 1223–1237. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Li, Z.; Zhuang, S.; Qi, S.; Li, L.; Zhou, J.; Zhang, W.; Zhao, Y. Melatonin alleviates inflammation-induced apoptosis in human umbilical vein endothelial cells via suppression of Ca2+-XO-ROS-Drp1-mitochondrial fission axis by activation of AMPK/SERCA2a pathway. Cell Stress Chaperones 2018, 23, 281–293. [Google Scholar] [CrossRef]
- Hong, Q.; Qi, K.; Feng, Z.; Huang, Z.; Cui, S.; Wang, L.; Fu, B.; Ding, R.; Yang, J.; Chen, X.; et al. Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2+ exchanger-mediated mitochondrial calcium overload. Cell Calcium 2012, 51, 402–410. [Google Scholar] [CrossRef]
- Arruda, A.P.; Hotamisligil, G.S. Calcium Homeostasis and Organelle Function in the Pathogenesis of Obesity and Diabetes. Cell Metab. 2015, 22, 381–397. [Google Scholar] [CrossRef] [Green Version]
- Petersen, O.H.; Tepikin, A.V.; Gerasimenko, J.V.; Gerasimenko, O.V.; Sutton, R.; Criddle, D.N. Fatty acids, alcohol and fatty acid ethyl esters: Toxic Ca2+ signal generation and pancreatitis. Cell Calcium 2009, 45, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Treviño, S.; Waalkes, M.P.; Flores Hernández, J.A.; León-Chavez, B.A.; Aguilar-Alonso, P.; Brambila, E. Chronic cadmium exposure in rats produces pancreatic impairment and insulin resistance in multiple peripheral tissues. Arch. Biochem. Biophys. 2015, 583, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Raqeeb, A.; Torres-Jácome, J.; Guzman-Silva, A.; Guerra, G.; Tanzi, F.; Moccia, F. The Mechanism of Injury-Induced Intracellular Calcium Concentration Oscillations in the Endothelium of Excised Rat Aorta. J. Vasc. Res. 2012, 49, 65–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Somatic Parameters | LZDF (n = 11) | OZDF (n = 14) |
| Weight (g) | 309.6 ± 6.03 | 529 ± 8.16 * |
| Length (cm) | 22.41 ± 0.29 | 23.5 ± 0.33 |
| Abdominal circumference (cm) | 13.21 ± 0.29 | 17.85 ± 0.36 * |
| BMI | 0.59 ± 0.009 | 0.93 ± 0.016 * |
| Epididymal fat (g) | 3.32 ± 0.12 | 15.71 ± 0.62 * |
| Biochemical Parameters | LZDF (n = 5) | OZDF (n = 5) |
| Total Cholesterol (mg/dL) | 90.83 ± 12.22 | 133 ± 11.82 * |
| HDL-C (mg/dL) | 61.6 ± 3.02 | 72.06 ± 8.22 |
| LDL-C (mg/dL) | 26.48 ± 12.09 | 35.64 ± 13.26 |
| VLDL (mg/dL) | 11.53 ± 3.62 | 34.53 ± 3.95 * |
| Triglycerides (mg/dL) | 42 ± 10.35 | 186.1 ± 23.04 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berra-Romani, R.; Guzmán-Silva, A.; Vargaz-Guadarrama, A.; Flores-Alonso, J.C.; Alonso-Romero, J.; Treviño, S.; Sánchez-Gómez, J.; Coyotl-Santiago, N.; García-Carrasco, M.; Moccia, F. Type 2 Diabetes Alters Intracellular Ca2+ Handling in Native Endothelium of Excised Rat Aorta. Int. J. Mol. Sci. 2020, 21, 250. https://doi.org/10.3390/ijms21010250
Berra-Romani R, Guzmán-Silva A, Vargaz-Guadarrama A, Flores-Alonso JC, Alonso-Romero J, Treviño S, Sánchez-Gómez J, Coyotl-Santiago N, García-Carrasco M, Moccia F. Type 2 Diabetes Alters Intracellular Ca2+ Handling in Native Endothelium of Excised Rat Aorta. International Journal of Molecular Sciences. 2020; 21(1):250. https://doi.org/10.3390/ijms21010250
Chicago/Turabian StyleBerra-Romani, Roberto, Alejandro Guzmán-Silva, Ajelet Vargaz-Guadarrama, Juan Carlos Flores-Alonso, José Alonso-Romero, Samuel Treviño, Josué Sánchez-Gómez, Nayeli Coyotl-Santiago, Mario García-Carrasco, and Francesco Moccia. 2020. "Type 2 Diabetes Alters Intracellular Ca2+ Handling in Native Endothelium of Excised Rat Aorta" International Journal of Molecular Sciences 21, no. 1: 250. https://doi.org/10.3390/ijms21010250
APA StyleBerra-Romani, R., Guzmán-Silva, A., Vargaz-Guadarrama, A., Flores-Alonso, J. C., Alonso-Romero, J., Treviño, S., Sánchez-Gómez, J., Coyotl-Santiago, N., García-Carrasco, M., & Moccia, F. (2020). Type 2 Diabetes Alters Intracellular Ca2+ Handling in Native Endothelium of Excised Rat Aorta. International Journal of Molecular Sciences, 21(1), 250. https://doi.org/10.3390/ijms21010250