β-Galactosylceramidase Deficiency Causes Bone Marrow Vascular Defects in an Animal Model of Krabbe Disease


 ,
,  and
and 
Abstract
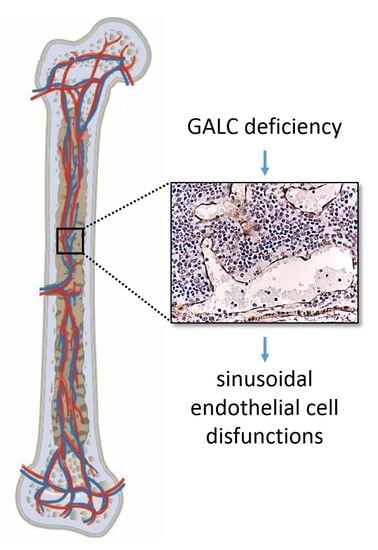
:
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Immunohistochemistry
4.3. Two-Photon Microscopy
4.3.1. Femurs Collection and Bone Clearing
4.3.2. Bone Immunostaining
4.3.3. Image Acquisition and Processing
4.4. Quantitative RT-PCR (RT-qPCR) Analysis
4.5. HUVEC Immunofluorescence Analysis
4.6. ELISA
4.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| KD | Krabbe disease |
| CNS | central nervous system |
| SEC | sinusoidal endothelial cell |
| GALC | β-galactosylceramidase |
| HSC | hematopoietic stem cell |
| HSCT | HSC transplantation |
| BM | bone marrow |
| VEGFR2 | vascular endothelial growth factor receptor-2 |
References
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta 2009, 1793, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Suzuki, Y. Globoid cell leucodystrophy (Krabbe’s disease): Deficiency of galactocerebroside beta-galactosidase. Proc. Natl. Acad. Sci. USA 1970, 66, 302–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K. Twenty five years of the “psychosine hypothesis”: A personal perspective of its history and present status. Neurochem. Res. 1998, 23, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Igisu, H.; Suzuki, K. Progressive accumulation of toxic metabolite in a genetic leukodystrophy. Science 1984, 224, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K. Globoid cell leukodystrophy (Krabbe’s disease): Update. J. Child. Neurol. 2003, 18, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Loonen, M.C.; Van Diggelen, O.P.; Janse, H.C.; Kleijer, W.J.; Arts, W.F. Late-onset globoid cell leucodystrophy (Krabbe’s disease). Clinical and genetic delineation of two forms and their relation to the early-infantile form. Neuropediatrics 1985, 16, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Wenger, D.A.; Rafi, M.A.; Luzi, P.; Datto, J.; Costantino-Ceccarini, E. Krabbe disease: Genetic aspects and progress toward therapy. Mol. Genet. Metab. 2000, 70, 1–9. [Google Scholar] [CrossRef]
- Sakai, N. Pathogenesis of leukodystrophy for Krabbe disease: Molecular mechanism and clinical treatment. Brain Dev. 2009, 31, 485–487. [Google Scholar] [CrossRef]
- Escolar, M.L.; Poe, M.D.; Provenzale, J.M.; Richards, K.C.; Allison, J.; Wood, S.; Wenger, D.A.; Pietryga, D.; Wall, D.; Champagne, M.; et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N. Engl. J. Med. 2005, 352, 2069–2081. [Google Scholar] [CrossRef] [Green Version]
- Escolar, M.L.; Poe, M.D.; Martin, H.R.; Kurtzberg, J. A staging system for infantile Krabbe disease to predict outcome after unrelated umbilical cord blood transplantation. Pediatrics 2006, 118, e879–e889. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K. The twitcher mouse. A model of human globoid cell leukodystrophy (krabbe’s disease). Am. J. Pathol. 1983, 111, 394–397. [Google Scholar] [PubMed]
- Suzuki, K. The twitcher mouse: A model for Krabbe disease and for experimental therapies. Brain Pathol. 1995, 5, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Visigalli, I.; Ungari, S.; Martino, S.; Park, H.; Cesani, M.; Gentner, B.; Sergi Sergi, L.; Orlacchio, A.; Naldini, L.; Biffi, A. The galactocerebrosidase enzyme contributes to the maintenance of a functional hematopoietic stem cell niche. Blood 2010, 116, 1857–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Q.; Frenette, P.S. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018, 48, 632–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, A.T.; Butler, J.M.; Nolan, D.J.; Kranz, A.; Iida, K.; Kobayashi, M.; Kopp, H.G.; Shido, K.; Petit, I.; Yanger, K.; et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell 2009, 4, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belleri, M.; Presta, M. Endothelial cell dysfunction in globoid cell leukodystrophy. J. Neurosci. Res. 2016, 94, 1359–1367. [Google Scholar] [CrossRef]
- Giacomini, A.; Ackermann, M.; Belleri, M.; Coltrini, D.; Nico, B.; Ribatti, D.; Konerding, M.A.; Presta, M.; Righi, M. Brain angioarchitecture and intussusceptive microvascular growth in a murine model of Krabbe disease. Angiogenesis 2015, 18, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Righi, M.; Belleri, M.; Presta, M.; Giacomini, A. Quantification of 3D Brain Microangioarchitectures in an Animal Model of Krabbe Disease. Int. J. Mol. Sci. 2019, 20, 2384. [Google Scholar] [CrossRef] [Green Version]
- Belleri, M.; Ronca, R.; Coltrini, D.; Nico, B.; Ribatti, D.; Poliani, P.L.; Giacomini, A.; Alessi, P.; Marchesini, S.; Santos, M.B.; et al. Inhibition of angiogenesis by beta-galactosylceramidase deficiency in globoid cell leukodystrophy. Brain 2013, 136, 2859–2875. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Shinoda, H.; Goto, I.; Yamanaka, T.; Suzuki, Y. Globoid cell leukodystrophy is a generalized galactosylsphingosine (psychosine) storage disease. Biochem. Biophys. Res. Commun. 1987, 144, 41–46. [Google Scholar] [CrossRef]
- Whitfield, P.D.; Sharp, P.C.; Taylor, R.; Meikle, P. Quantification of galactosylsphingosine in the twitcher mouse using electrospray ionization-tandem mass spectrometry. J. Lipid Res. 2001, 42, 2092–2095. [Google Scholar] [PubMed]
- Contreras, M.A.; Ries, W.L.; Shanmugarajan, S.; Arboleda, G.; Singh, I.; Singh, A.K. Factors that affect postnatal bone growth retardation in the twitcher murine model of Krabbe disease. Biochim. Biophys. Acta 2010, 1802, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Contreras, M.A.; Haq, E.; Uto, T.; Singh, I.; Singh, A.K. Psychosine-induced alterations in peroxisomes of twitcher mouse liver. Arch. Biochem. Biophys. 2008, 477, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galbiati, F.; Basso, V.; Cantuti, L.; Givogri, M.I.; Lopez-Rosas, A.; Perez, N.; Vasu, C.; Cao, H.; van Breemen, R.; Mondino, A.; et al. Autonomic denervation of lymphoid organs leads to epigenetic immune atrophy in a mouse model of Krabbe disease. J. Neurosci. 2007, 27, 13730–13738. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Bao, S.D. Roles of main pro- and anti-angiogenic factors in tumor angiogenesis. World J. Gastroenterol. 2004, 10, 463–470. [Google Scholar] [CrossRef]
- Potter, G.B.; Petryniak, M.A. Neuroimmune mechanisms in Krabbe’s disease. J. Neurosci. Res. 2016, 94, 1341–1348. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol. Neurobiol. 2000, 20, 131–147. [Google Scholar] [CrossRef]
- Kanazawa, T.; Nakamura, S.; Momoi, M.; Yamaji, T.; Takematsu, H.; Yano, H.; Sabe, H.; Yamamoto, A.; Kawasaki, T.; Kozutsumi, Y. Inhibition of cytokinesis by a lipid metabolite, psychosine. J. Cell Biol. 2000, 149, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Kozutsumi, Y.; Kanazawa, T.; Sun, Y.; Yamaji, T.; Yamamoto, H.; Takematsu, H. Sphingolipids involved in the induction of multinuclear cell formation. Biochim. Biophys. Acta 2002, 1582, 138–143. [Google Scholar] [CrossRef]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.; Seppanen, E.J.; Rodero, M.P.; Wong, H.Y.; Donovan, P.; Neufeld, Z.; Fisk, N.M.; Francois, M.; Khosrotehrani, K. Functional Definition of Progenitors Versus Mature Endothelial Cells Reveals Key SoxF-Dependent Differentiation Process. Circulation 2017, 135, 786–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef] [PubMed]
- Maddaluno, L.; Rudini, N.; Cuttano, R.; Bravi, L.; Giampietro, C.; Corada, M.; Ferrarini, L.; Orsenigo, F.; Papa, E.; Boulday, G.; et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 2013, 498, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Sivaraj, K.K.; Adams, R.H. Blood vessel formation and function in bone. Development 2016, 143, 2706–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, N.; Inui, K.; Tatsumi, N.; Fukushima, H.; Nishigaki, T.; Taniike, M.; Nishimoto, J.; Tsukamoto, H.; Yanagihara, I.; Ozono, K.; et al. Molecular cloning and expression of cDNA for murine galactocerebrosidase and mutation analysis of the twitcher mouse, a model of Krabbe’s disease. J. Neurochem. 1996, 66, 1118–1124. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| Cd31 | 5′-CGGTTATGATGATGTTTCTGGA-3′ | 5′-AAGGGAGGACACTTCCACTTCT-3′ |
| Vegfr2 | 5′-AGATGCCCGACTCCCTTT-3′ | 5′-TTTCCCAGAGCAACACACC-3′ |
| Vegfa | 55′-ACCTCCACCATGCCAAGT-3′ | 5′-TCAATCGGACGGCAGTAG-3′ |
| Fgf2 | 5′-CCTTCCCACCAGGCCACTTAA-3’ | 5′-GGTCCGTTTTGGATCCGAGTTT-3’ |
| Angpt1 | 5′-TCATGCTAACAGGAGGTTGGT-3′ | 5′-ATGGTGGTGGAACGTAAGGA-3′ |
| Angpt2 | 5′-GCTGGGCAATGAGTTTGTCT-3′ | 5′-CCTGGTTGGCTGATGCTACT-3′ |
| Il1α | 5′-CAGTTCTGCCATTGACCATC-3′ | 5′-GAATCTTCCCGTTGCTTGAC-3′ |
| Tnfα | 5′-GCCTCTTCTCATTCCTGCTT-3′ | 5′-TGATCTGAGTGTGAGGGTCTG-3′ |
| Tgfβ1 | 5′-TTGCTTCAGCTCCACAGAGA-3′ | 5′-TACTGTGTGTCCAGGCTCCA-3′ |
| Gapdh | 5′-GAAGGTCGGTGTGAACGGATT-3′ | 5′-TGACTGTGCCGTTGAATTTG-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belleri, M.; Coltrini, D.; Righi, M.; Ravelli, C.; Taranto, S.; Chiodelli, P.; Mitola, S.; Presta, M.; Giacomini, A. β-Galactosylceramidase Deficiency Causes Bone Marrow Vascular Defects in an Animal Model of Krabbe Disease. Int. J. Mol. Sci. 2020, 21, 251. https://doi.org/10.3390/ijms21010251
Belleri M, Coltrini D, Righi M, Ravelli C, Taranto S, Chiodelli P, Mitola S, Presta M, Giacomini A. β-Galactosylceramidase Deficiency Causes Bone Marrow Vascular Defects in an Animal Model of Krabbe Disease. International Journal of Molecular Sciences. 2020; 21(1):251. https://doi.org/10.3390/ijms21010251
Chicago/Turabian StyleBelleri, Mirella, Daniela Coltrini, Marco Righi, Cosetta Ravelli, Sara Taranto, Paola Chiodelli, Stefania Mitola, Marco Presta, and Arianna Giacomini. 2020. "β-Galactosylceramidase Deficiency Causes Bone Marrow Vascular Defects in an Animal Model of Krabbe Disease" International Journal of Molecular Sciences 21, no. 1: 251. https://doi.org/10.3390/ijms21010251
APA StyleBelleri, M., Coltrini, D., Righi, M., Ravelli, C., Taranto, S., Chiodelli, P., Mitola, S., Presta, M., & Giacomini, A. (2020). β-Galactosylceramidase Deficiency Causes Bone Marrow Vascular Defects in an Animal Model of Krabbe Disease. International Journal of Molecular Sciences, 21(1), 251. https://doi.org/10.3390/ijms21010251