Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
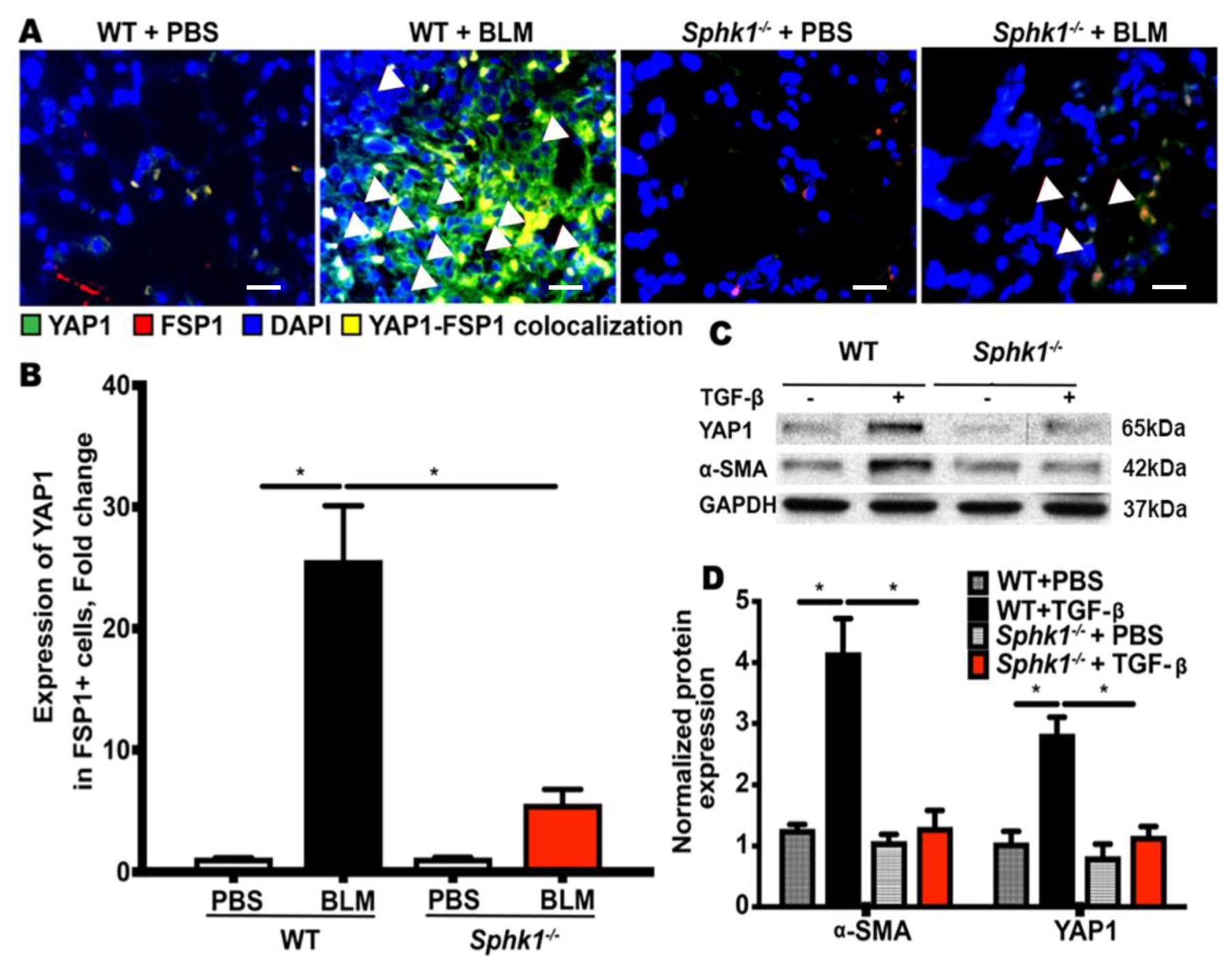
2.1. Genetic Deletion of Sphk1 in Fibroblasts and Alveolar Epithelial Cells Protects Mice against Bleomycin-Induced Lung Fibrosis
2.2. Genetic Deletion of Sphk1 in Fibroblasts Reduces Bleomycin- and TGF-β-Induced YAP1 Expression
2.3. SPHK1 Activity is Essential for Bleomycin- and TGF-β-Mediated YAP1 Expression in Lung Fibroblasts
2.4. Inhibition of SPHK1 by PF543 Attenuates Bleomycin- and TGF-β-Induced Mitochondrial ROS Generation and Expression of FN and α-SMA in Lung Fibroblasts
2.5. Inhibition of SPHK1 Activity with PF543 Attenuates Bleomycin- and TGF-β-Mediated YAP1 Translocation to The Nucleus
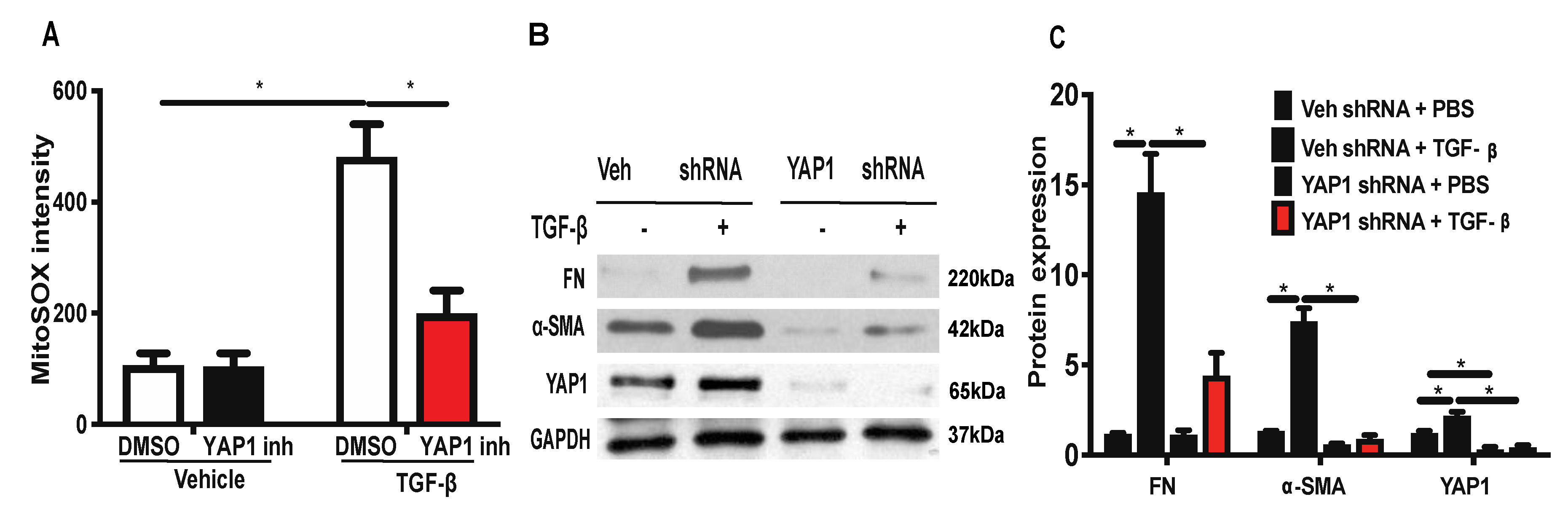
2.6. Inhibition or Downregulation of YAP1 Reduces TGF-β-Induced Mitochondrial ROS Generation and Expression of FN and α-SMA in Lung Fibroblasts
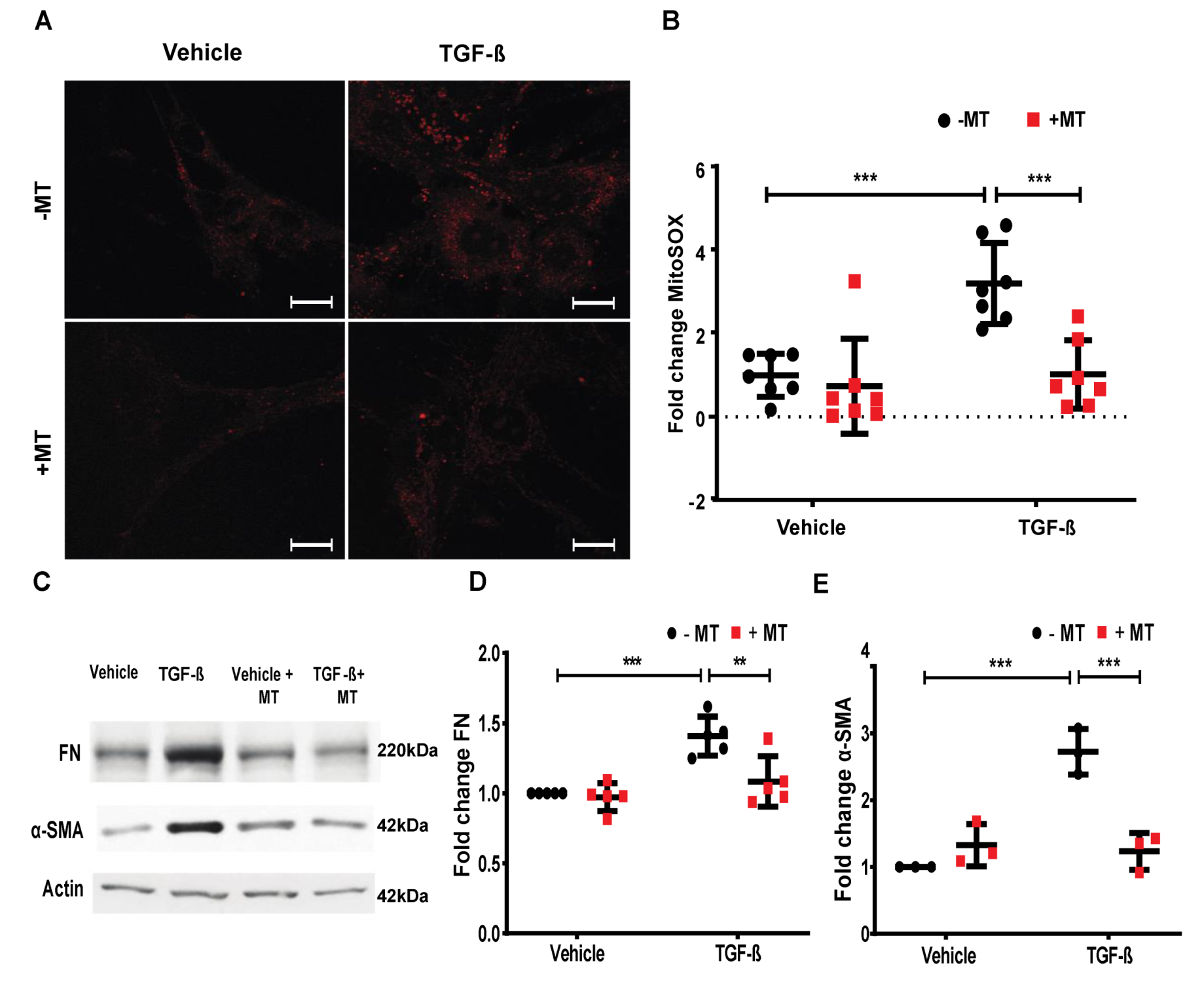
2.7. Mitochondrial ROS is Essential for TGF-β-Induced Expression of FN and α-SMA in Lung Fibroblasts
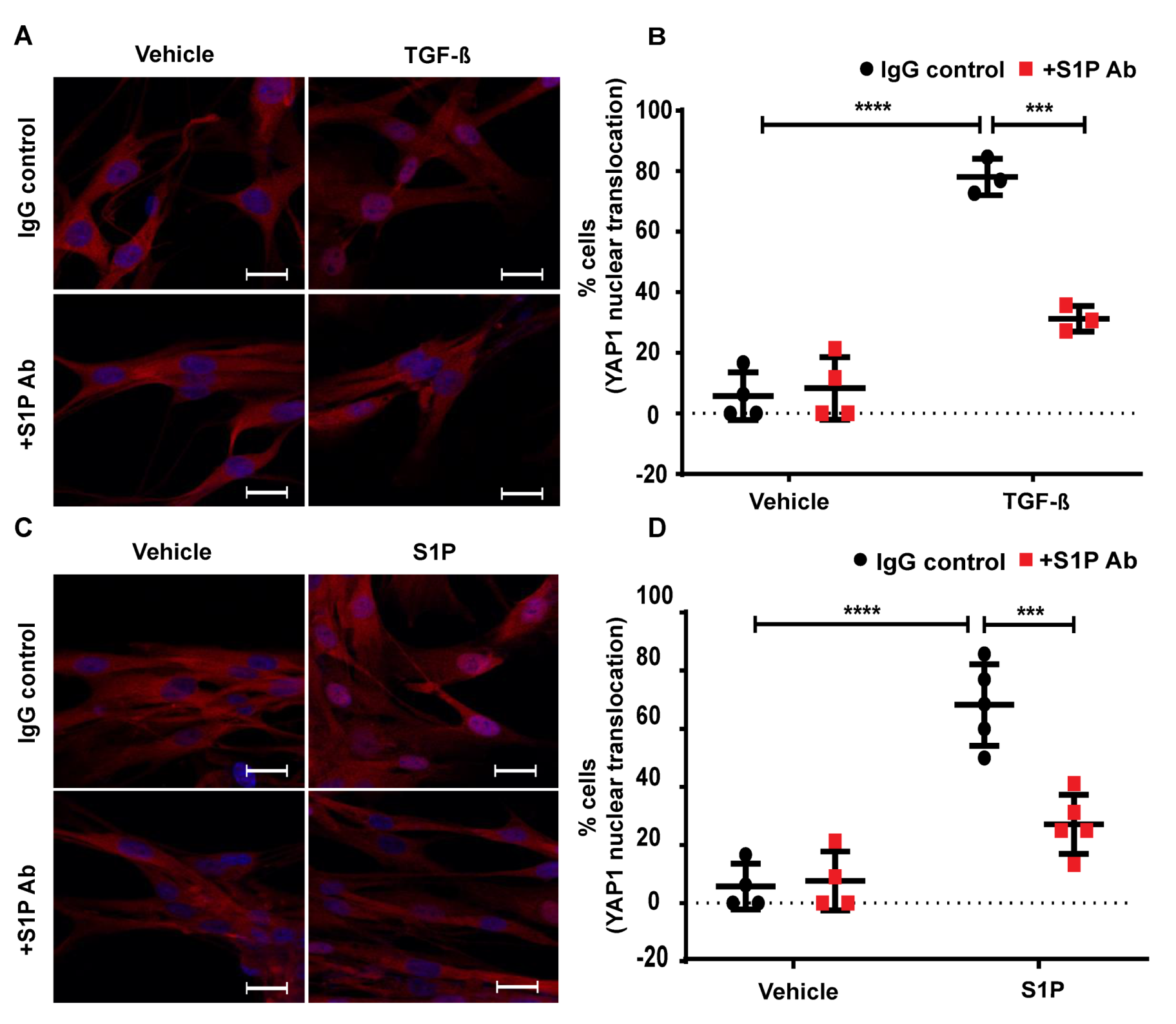
2.8. TGF-β- and S1P-Induced mtROS Production in HLFs is Attenuated with S1P-Antibody
2.9. TGF-β-Induced YAP1 Nuclear Translocation in Lung Fibroblasts is Attenuated by S1P Antibody
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Mice
4.3. Histopathological Analysis of Pulmonary Fibrosis
4.4. Collagen Content in Lung Tissue
4.5. Isolation of Primary Fibroblasts from Mouse Lungs and Cell Culture
4.6. Immunofluorescence Microscopy
4.7. Western Blot
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SPHK1 | Sphingosine kinase 1 |
| S1P | Sphingosine-1-phosphate |
| YAP | Yes-related protein |
| MLF | Mouse Lung Fibroblast |
| HLF | Human Lung Fibroblast |
| FN | Fibronectin |
| α-SMA | Alpha-smooth muscle actin |
| mtROS | Mitochondrial reactive oxygen species |
| IPF | Idiopathic pulmonary fibrosis |
| PF | Pulmonary fibrosis |
| AEC | Alveolar Epithelial Cell |
References
- King, T.E. Update in pulmonary medicine. Ann. Intern. Med. 1998, 129, 806–812. [Google Scholar] [CrossRef]
- Selman, M.; King, T.E.; Pardo, A. American Thoracic Society; European Respiratory Society; American College of Chest Physicians Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.L.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, M.; Liu, Y.; Ma, S.; Yu, Z. Current advances in idiopathic pulmonary fibrosis: The pathogenesis, therapeutic strategies and candidate molecules. Future Med. Chem. 2019, 11, 2595–2620. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.F.; Raghu, G. Antifibrotic therapy for fibrotic lung disease beyond idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2019, 28, 190022. [Google Scholar] [CrossRef]
- George, P.M.; Patterson, C.M.; Reed, A.K.; Thillai, M. Lung transplantation for idiopathic pulmonary fibrosis. Lancet Respir. Med. 2019, 7, 271–282. [Google Scholar] [CrossRef]
- Wuyts, W.A.; Wijsenbeek, M.; Bondue, B.; Bouros, D.; Bresser, P.; Robalo Cordeiro, C.; Hilberg, O.; Magnusson, J.; Manali, E.D.; Morais, A.; et al. Idiopathic Pulmonary Fibrosis: Best Practice in Monitoring and Managing a Relentless Fibrotic Disease. Respiration 2019, 99, 1–10. [Google Scholar] [CrossRef]
- Maher, T.M.; Evans, I.C.; Bottoms, S.E.; Mercer, P.F.; Thorley, A.J.; Nicholson, A.G.; Laurent, G.J.; Tetley, T.D.; Chambers, R.C.; McAnulty, R.J. Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Wei, B.; Cai, L.; Sun, D.; Wang, Y.; Wang, C.; Chai, X.; Xie, F.; Su, M.; Ding, F.; Liu, J.; et al. Microsomal prostaglandin E synthase-1 deficiency exacerbates pulmonary fibrosis induced by BLM in mice. Molecules 2014, 19, 4967–4985. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Sheng, W.; Michkov, A.; Sriarm, K.; Sun, R.; Dvorkin-Gheva, A.; Insel, P.A.; Janssen, L.J. Prostaglandin E2 inhibits profibrotic function of human pulmonary fibroblasts by disrupting Ca2+ signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L810–L821. [Google Scholar] [CrossRef]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Shea, B.S.; Tager, A.M. Role of the lysophospholipid mediators lysophosphatidic acid and sphingosine 1-phosphate in lung fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninou, I.; Kaffe, E.; Müller, S.; Budd, D.C.; Stevenson, C.S.; Ullmer, C.; Aidinis, V. Pharmacologic targeting of the ATX/LPA axis attenuates BLM-induced pulmonary fibrosis. Pulm. Pharmacol. Ther. 2018, 52, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Funke, M.; Zhao, Z.; Xu, Y.; Chun, J.; Tager, A.M. The lysophosphatidic acid receptor LPA1 promotes epithelial cell apoptosis after lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 46, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.S.; Fu, P.; Patel, P.; Harijith, A.; Sun, T.; Zhao, Y.; Garcia, J.G.N.; Chun, J.; Natarajan, V. Lysophosphatidic acid receptor-2 deficiency confers protection against BLM-induced lung injury and fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 2013, 49, 912–922. [Google Scholar] [CrossRef] [Green Version]
- Takuwa, Y.; Du, W.; Qi, X.; Okamoto, Y.; Takuwa, N.; Yoshioka, K. Roles of sphingosine-1-phosphate signaling in angiogenesis. World J. Biol. Chem. 2010, 1, 298–306. [Google Scholar] [CrossRef]
- Pyne, N.J.; Dubois, G.; Pyne, S. Role of sphingosine 1-phosphate and lysophosphatidic acid in fibrosis. Biochim. Biophys. Acta 2013, 1831, 228–238. [Google Scholar] [CrossRef]
- Huang, L.S.; Natarajan, V. Sphingolipids in pulmonary fibrosis. Adv. Biol. Regul. 2015, 57, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Wadgaonkar, R.; Patel, V.; Grinkina, N.; Romano, C.; Liu, J.; Zhao, Y.; Sammani, S.; Garcia, J.G.N.; Natarajan, V. Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L603–L613. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, V.; Dudek, S.M.; Jacobson, J.R.; Moreno-Vinasco, L.; Huang, L.S.; Abassi, T.; Mathew, B.; Zhao, Y.; Wang, L.; Bittman, R.; et al. Sphingosine-1-phosphate, FTY720, and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 6–17. [Google Scholar] [CrossRef] [Green Version]
- Ebenezer, D.L.; Fu, P.; Natarajan, V. Targeting sphingosine-1-phosphate signaling in lung diseases. Pharmacol. Ther. 2016, 168, 143–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Gorshkova, I.A.; Berdyshev, E.; He, D.; Fu, P.; Ma, W.; Su, Y.; Usatyuk, P.V.; Pendyala, S.; Oskouian, B.; et al. Protection of LPS-induced murine acute lung injury by sphingosine-1-phosphate lyase suppression. Am. J. Respir. Cell Mol. Biol. 2011, 45, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.S.; Berdyshev, E.V.; Tran, J.T.; Xie, L.; Chen, J.; Ebenezer, D.L.; Mathew, B.; Gorshkova, I.; Zhang, W.; Reddy, S.P.; et al. Sphingosine-1-phosphate lyase is an endogenous suppressor of pulmonary fibrosis: Role of S1P signalling and autophagy. Thorax 2015, 70, 1138–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, J.D. Fifty years of lyase and a moment of truth: Sphingosine phosphate lyase from discovery to disease. J. Lipid Res. 2019, 60, 456–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brindley, D.N. Lipid phosphate phosphatases and related proteins: Signaling functions in development, cell division, and cancer. J. Cell. Biochem. 2004, 92, 900–912. [Google Scholar] [CrossRef]
- Huang, L.S.; Berdyshev, E.; Mathew, B.; Fu, P.; Gorshkova, I.A.; He, D.; Ma, W.; Noth, I.; Ma, S.-F.; Pendyala, S.; et al. Targeting sphingosine kinase 1 attenuates BLM-induced pulmonary fibrosis. FASEB J. 2013, 27, 1749–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luzina, I.G.; Salcedo, M.V.; Rojas-Peña, M.L.; Wyman, A.E.; Galvin, J.R.; Sachdeva, A.; Clerman, A.; Kim, J.; Franks, T.J.; Britt, E.J.; et al. Transcriptomic evidence of immune activation in macroscopically normal-appearing and scarred lung tissues in idiopathic pulmonary fibrosis. Cell. Immunol. 2018, 325, 1–13. [Google Scholar] [CrossRef]
- Hirahara, K.; Aoki, A.; Morimoto, Y.; Kiuchi, M.; Okano, M.; Nakayama, T. The immunopathology of lung fibrosis: Amphiregulin-producing pathogenic memory T helper-2 cells control the airway fibrotic responses by inducing eosinophils to secrete osteopontin. Semin. Immunopathol. 2019, 41, 339–348. [Google Scholar] [CrossRef]
- Morishima, Y.; Nomura, A.; Uchida, Y.; Noguchi, Y.; Sakamoto, T.; Ishii, Y.; Goto, Y.; Masuyama, K.; Zhang, M.J.; Hirano, K.; et al. Triggering the induction of myofibroblast and fibrogenesis by airway epithelial shedding. Am. J. Respir. Cell Mol. Biol. 2001, 24, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Tager, A.M. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 911–921. [Google Scholar] [CrossRef] [Green Version]
- Suryadevara, V.; Huang, L.; Kim, S.-J.; Cheresh, P.; Shaaya, M.; Bandela, M.; Fu, P.; Feghali-Bostwick, C.; Di Paolo, G.; Kamp, D.W.; et al. Role of phospholipase D in BLM-induced mitochondrial reactive oxygen species generation, mitochondrial DNA damage, and pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L175–L187. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Wu, L.; Deng, Z.; Huo, Y.; Cheng, Y. Emerging roles of YAP/TAZ in lung physiology and diseases. Life Sci. 2018, 214, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Jorgenson, A.J.; Choi, K.M.; Sicard, D.; Smith, K.M.J.; Hiemer, S.E.; Varelas, X.; Tschumperlin, D.J. TAZ activation drives fibroblast spheroid growth, expression of profibrotic paracrine signals, and context-dependent ECM gene expression. Am. J. Physiol. Cell Physiol. 2017, 312, C277–C285. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Saito, A.; Mikami, Y.; Urushiyama, H.; Horie, M.; Matsuzaki, H.; Takeshima, H.; Makita, K.; Miyashita, N.; Mitani, A.; et al. TAZ contributes to pulmonary fibrosis by activating profibrotic functions of lung fibroblasts. Sci. Rep. 2017, 7, 42595. [Google Scholar] [CrossRef]
- Gokey, J.J.; Sridharan, A.; Xu, Y.; Green, J.; Carraro, G.; Stripp, B.R.; Perl, A.-K.T.; Whitsett, J.A. Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. JCI Insight 2018, 3, e98738. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.; Yang, J.; DeRan, M.; Wu, C.; Su, A.I.; Bonamy, G.M.C.; Liu, J.; Peters, E.C.; Wu, X. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem. Biol. 2012, 19, 955–962. [Google Scholar] [CrossRef] [Green Version]
- Kemppainen, K.; Wentus, N.; Lassila, T.; Laiho, A.; Törnquist, K. Sphingosylphosphorylcholine regulates the Hippo signaling pathway in a dual manner. Cell. Signal. 2016, 28, 1894–1903. [Google Scholar] [CrossRef]
- Schnute, M.E.; McReynolds, M.D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J.W.; Hall, T.; Chrencik, J.; Kraus, M.; Cronin, C.N.; et al. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 2012, 444, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Malsin, E.S.; Kamp, D.W. The mitochondria in lung fibrosis: Friend or foe? Transl. Res. 2018, 202, 1–23. [Google Scholar] [CrossRef]
- Harijith, A.; Pendyala, S.; Ebenezer, D.L.; Ha, A.W.; Fu, P.; Wang, Y.-T.; Ma, K.; Toth, P.T.; Berdyshev, E.V.; Kanteti, P.; et al. Hyperoxia-induced p47phox activation and ROS generation is mediated through S1P transporter Spns2, and S1P/S1P1&2 signaling axis in lung endothelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L337–L351. [Google Scholar] [PubMed] [Green Version]
- Kim, S.; Sieburth, D. Sphingosine Kinase Activates the Mitochondrial Unfolded Protein Response and Is Targeted to Mitochondria by Stress. Cell Rep. 2018, 24, 2932–2945.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Huang, J.; Wang, X.; Yuan, J.; Li, X.; Feng, L.; Park, J.-I.; Chen, J. PTPN14 is required for the density-dependent control of YAP1. Genes Dev. 2012, 26, 1959–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komuro, A.; Nagai, M.; Navin, N.E.; Sudol, M. WW domain-containing protein YAP associates with ErbB-4 and acts as a co-transcriptional activator for the carboxyl-terminal fragment of ErbB-4 that translocates to the nucleus. J. Biol. Chem. 2003, 278, 33334–33341. [Google Scholar] [CrossRef] [Green Version]
- Gibault, F.; Bailly, F.; Corvaisier, M.; Coevoet, M.; Huet, G.; Melnyk, P.; Cotelle, P. Molecular Features of the YAP Inhibitor Verteporfin: Synthesis of Hexasubstituted Dipyrrins as Potential Inhibitors of YAP/TAZ, the Downstream Effectors of the Hippo Pathway. ChemMedChem 2017, 12, 954–961. [Google Scholar] [CrossRef]
- Li, Y.; Wang, S.; Wei, X.; Zhang, S.; Song, Z.; Chen, X.; Zhang, J. Role of inhibitor of yes-associated protein 1 in triple-negative breast cancer with taxol-based chemoresistance. Cancer Sci. 2019, 110, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Kono, Y.; Nishiuma, T.; Nishimura, Y.; Kotani, Y.; Okada, T.; Nakamura, S.-I.; Yokoyama, M. Sphingosine kinase 1 regulates differentiation of human and mouse lung fibroblasts mediated by TGF-β 1. Am. J. Respir. Cell Mol. Biol. 2007, 37, 395–404. [Google Scholar] [CrossRef]
- Cencetti, F.; Bernacchioni, C.; Nincheri, P.; Donati, C.; Bruni, P. Transforming growth factor-beta1 induces transdifferentiation of myoblasts into myofibroblasts via up-regulation of sphingosine kinase-1/S1P3 axis. Mol. Biol. Cell 2010, 21, 1111–1124. [Google Scholar] [CrossRef] [Green Version]
- Milara, J.; Navarro, R.; Juan, G.; Peiró, T.; Serrano, A.; Ramón, M.; Morcillo, E.; Cortijo, J. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012, 67, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Gorshkova, I.; Zhou, T.; Mathew, B.; Jacobson, J.R.; Takekoshi, D.; Bhattacharya, P.; Smith, B.; Aydogan, B.; Weichselbaum, R.R.; Natarajan, V.; et al. Inhibition of serine palmitoyltransferase delays the onset of radiation-induced pulmonary fibrosis through the negative regulation of sphingosine kinase-1 expression. J. Lipid Res. 2012, 53, 1553–1568. [Google Scholar] [CrossRef] [Green Version]
- Khalil, N.; O’Connor, R.N.; Unruh, H.W.; Warren, P.W.; Flanders, K.C.; Kemp, A.; Bereznay, O.H.; Greenberg, A.H. Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 1991, 5, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Limper, A.H.; Broekelmann, T.J.; Colby, T.V.; Malizia, G.; McDonald, J.A. Analysis of local mRNA expression for extracellular matrix proteins and growth factors using in situ hybridization in fibroproliferative lung disorders. Chest 1991, 99, 55S–56S. [Google Scholar] [CrossRef]
- Bartram, U.; Speer, C.P. The role of transforming growth factor beta in lung development and disease. Chest 2004, 125, 754–765. [Google Scholar] [CrossRef]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.-K.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Zhai, C.; Pan, Y.; Zhu, Y.; Shi, W.; Wang, J.; Yan, X.; Su, X.; Song, Y.; Gao, L.; et al. Sphingosine-1-phosphate induces airway smooth muscle cell proliferation, migration, and contraction by modulating Hippo signaling effector YAP. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L609–L621. [Google Scholar] [CrossRef]
- Shen, Y.; Zhao, S.; Wang, S.; Pan, X.; Zhang, Y.; Xu, J.; Jiang, Y.; Li, H.; Zhang, Q.; Gao, J.; et al. S1P/S1PR3 axis promotes aerobic glycolysis by YAP/c-MYC/PGAM1 axis in osteosarcoma. EBioMedicine 2019, 40, 210–223. [Google Scholar] [CrossRef] [Green Version]
- Munro, D.; Treberg, J.R. A radical shift in perspective: Mitochondria as regulators of reactive oxygen species. J. Exp. Biol. 2017, 220, 1170–1180. [Google Scholar] [CrossRef] [Green Version]
- van der Vliet, A.; Janssen-Heininger, Y.M.W.; Anathy, V. Oxidative stress in chronic lung disease: From mitochondrial dysfunction to dysregulated redox signaling. Mol. Asp. Med. 2018, 63, 59–69. [Google Scholar] [CrossRef]
- Veith, C.; Boots, A.W.; Idris, M.; van Schooten, F.-J.; van der Vliet, A. Redox Imbalance in Idiopathic Pulmonary Fibrosis: A Role for Oxidant Cross-Talk Between NADPH Oxidase Enzymes and Mitochondria. Antioxid. Redox Signal. 2019, 31, 1092–1115. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, Z. The pathophysiological role of mitochondrial oxidative stress in lung diseases. J. Transl. Med. 2017, 15, 207. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, K.; Nakashima, N.; Inoshima, I.; Hagimoto, N.; Fujita, M.; Yoshimi, M.; Maeyama, T.; Hamada, N.; Watanabe, K.; Hara, N. Oxidative stress in lung epithelial cells from patients with idiopathic interstitial pneumonias. Eur. Respir. J. 2003, 21, 232–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamp, D.W.; Liu, G.; Cheresh, P.; Kim, S.-J.; Mueller, A.; Lam, A.P.; Trejo, H.; Williams, D.; Tulasiram, S.; Baker, M.; et al. Asbestos-induced alveolar epithelial cell apoptosis. The role of endoplasmic reticulum stress response. Am. J. Respir. Cell Mol. Biol. 2013, 49, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Cheresh, P.; Jablonski, R.P.; Morales-Nebreda, L.; Cheng, Y.; Hogan, E.; Yeldandi, A.; Chi, M.; Piseaux, R.; Ridge, K.; et al. Mitochondrial catalase overexpressed transgenic mice are protected against lung fibrosis in part via preventing alveolar epithelial cell mitochondrial DNA damage. Free Radic. Biol. Med. 2016, 101, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-J.; Cheresh, P.; Williams, D.; Cheng, Y.; Ridge, K.; Schumacker, P.T.; Weitzman, S.; Bohr, V.A.; Kamp, D.W. Mitochondria-targeted Ogg1 and aconitase-2 prevent oxidant-induced mitochondrial DNA damage in alveolar epithelial cells. J. Biol. Chem. 2014, 289, 6165–6176. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Dada, L.A.; Wu, M.; Kelly, A.; Trejo, H.; Zhou, Q.; Varga, J.; Sznajder, J.I. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1120–L1130. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.-S.; Lee, J.-H.; Hwang, S.-C.; Choi, K.S.; Yoon, G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 2005, 24, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Graham, K.A.; Kulawiec, M.; Owens, K.M.; Li, X.; Desouki, M.M.; Chandra, D.; Singh, K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 2010, 10, 223–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-M.; Kim, Y.-G.; Jeong, K.-H.; Lee, S.-H.; Lee, T.-W.; Ihm, C.-G.; Moon, J.-Y. Angiotensin II-induced mitochondrial Nox4 is a major endogenous source of oxidative stress in kidney tubular cells. PLoS ONE 2012, 7, e39739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, A.J.; Li, S.; Basu, U.; Tian, J.; Zimmerman, M.C. Mitochondrial-localized NADPH oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H19–H28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazziano, G.; Al Ghouleh, I.; Baust, J.; Shiva, S.; Champion, H.C.; Pagano, P.J. Nox-derived ROS are acutely activated in pressure overload pulmonary hypertension: Indications for a seminal role for mitochondrial Nox4. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H197–H205. [Google Scholar] [CrossRef] [Green Version]
- Das, R.; Xu, S.; Quan, X.; Nguyen, T.T.; Kong, I.D.; Chung, C.H.; Lee, E.Y.; Cha, S.-K.; Park, K.-S. Upregulation of mitochondrial Nox4 mediates TGF-β-induced apoptosis in cultured mouse podocytes. Am. J. Physiol. Renal Physiol. 2014, 306, F155–F167. [Google Scholar] [CrossRef]
- Hirschhäuser, C.; Bornbaum, J.; Reis, A.; Böhme, S.; Kaludercic, N.; Menabò, R.; Di Lisa, F.; Boengler, K.; Shah, A.M.; Schulz, R.; et al. NOX4 in Mitochondria: Yeast Two-Hybrid-Based Interaction with Complex I Without Relevance for Basal Reactive Oxygen Species? Antioxid. Redox Signal. 2015, 23, 1106–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canugovi, C.; Stevenson, M.D.; Vendrov, A.E.; Hayami, T.; Robidoux, J.; Xiao, H.; Zhang, Y.-Y.; Eitzman, D.T.; Runge, M.S.; Madamanchi, N.R. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. 2019, 26, 101288. [Google Scholar] [CrossRef] [PubMed]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurundkar, A.; Thannickal, V.J. Redox mechanisms in age-related lung fibrosis. Redox Biol. 2016, 9, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Ago, T.; Matsushima, S.; Kuroda, J.; Zablocki, D.; Kitazono, T.; Sadoshima, J. The NADPH oxidase Nox4 and aging in the heart. Aging (Albany NY) 2010, 2, 1012–1016. [Google Scholar] [CrossRef] [Green Version]
- McCrann, D.J.; Yang, D.; Chen, H.; Carroll, S.; Ravid, K. Upregulation of Nox4 in the aging vasculature and its association with smooth muscle cell polyploidy. Cell Cycle 2009, 8, 902–908. [Google Scholar] [CrossRef] [Green Version]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarman, E.R.; Khambata, V.S.; Cope, C.; Jones, P.; Roger, J.; Ye, L.Y.; Duggan, N.; Head, D.; Pearce, A.; Press, N.J.; et al. An inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am. J. Respir. Cell Mol. Biol. 2014, 50, 158–169. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Larson-Casey, J.L.; Davis, D.; Hanumanthu, V.S.; Longhini, A.L.F.; Thannickal, V.J.; Gu, L.; Carter, A.B. NOX4 modulates macrophage phenotype and mitochondrial biogenesis in asbestosis. JCI Insight 2019, 4, e126551. [Google Scholar] [CrossRef] [PubMed]
- Kozieł, R.; Pircher, H.; Kratochwil, M.; Lener, B.; Hermann, M.; Dencher, N.A.; Jansen-Dürr, P. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem. J. 2013, 452, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, M.A.; MacKinnon, A.C.; Ramachandran, P.; Dhaliwal, K.; Duffin, R.; Phythian-Adams, A.T.; van Rooijen, N.; Haslett, C.; Howie, S.E.; Simpson, A.J.; et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Kim, S.-J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Cell Physiol. 2017, 312, C749–C764. [Google Scholar] [CrossRef] [Green Version]












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, L.S.; Sudhadevi, T.; Fu, P.; Punathil-Kannan, P.-K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. Int. J. Mol. Sci. 2020, 21, 2064. https://doi.org/10.3390/ijms21062064
Huang LS, Sudhadevi T, Fu P, Punathil-Kannan P-K, Ebenezer DL, Ramchandran R, Putherickal V, Cheresh P, Zhou G, Ha AW, et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. International Journal of Molecular Sciences. 2020; 21(6):2064. https://doi.org/10.3390/ijms21062064
Chicago/Turabian StyleHuang, Long Shuang, Tara Sudhadevi, Panfeng Fu, Prasanth-Kumar Punathil-Kannan, David Lenin Ebenezer, Ramaswamy Ramchandran, Vijay Putherickal, Paul Cheresh, Guofei Zhou, Alison W. Ha, and et al. 2020. "Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts" International Journal of Molecular Sciences 21, no. 6: 2064. https://doi.org/10.3390/ijms21062064
APA StyleHuang, L. S., Sudhadevi, T., Fu, P., Punathil-Kannan, P. -K., Ebenezer, D. L., Ramchandran, R., Putherickal, V., Cheresh, P., Zhou, G., Ha, A. W., Harijith, A., Kamp, D. W., & Natarajan, V. (2020). Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. International Journal of Molecular Sciences, 21(6), 2064. https://doi.org/10.3390/ijms21062064