Sphingolipids in Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma: Ceramide Turnover

 ,
,  , and
, and 
Abstract
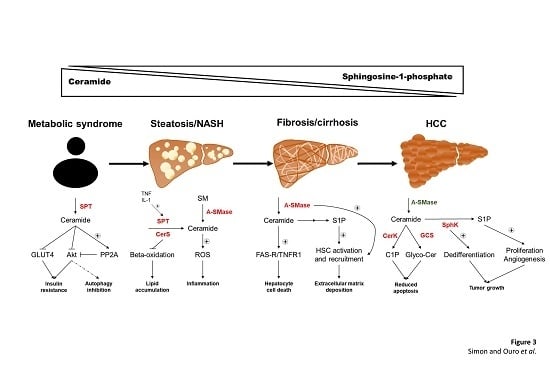
:
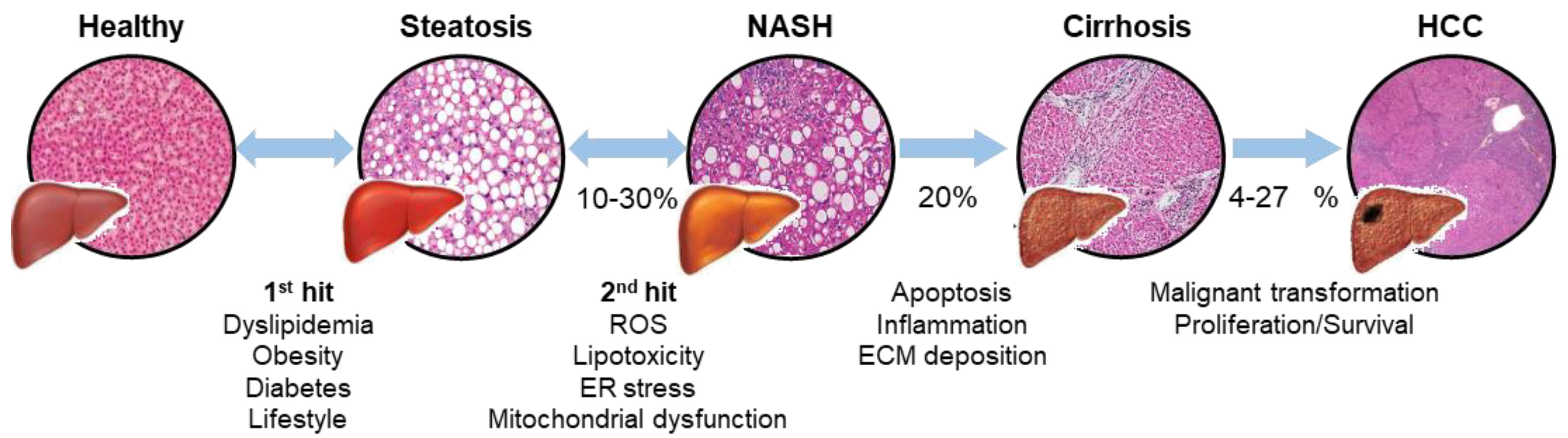
{kind=link}
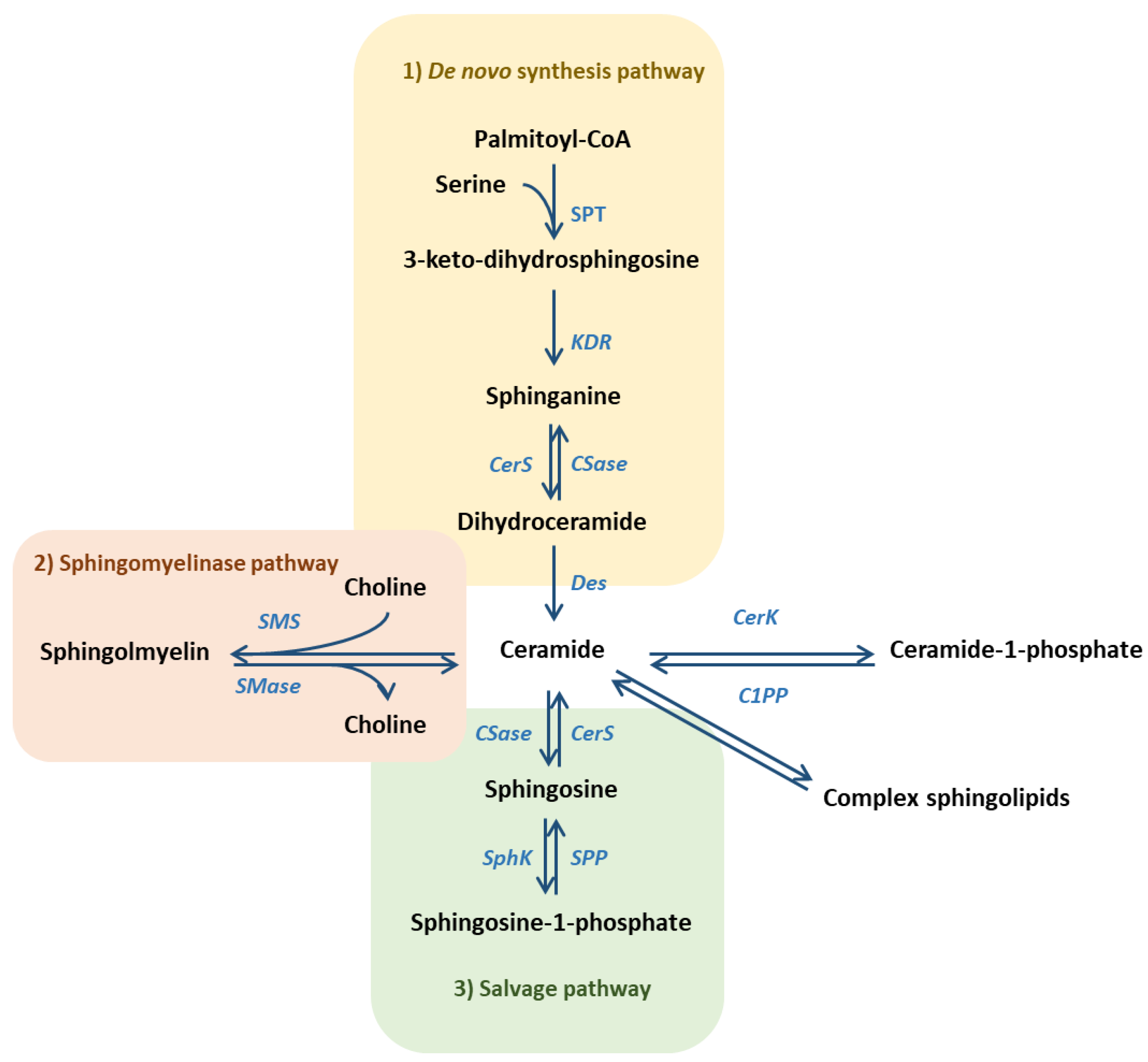
{kind=link}
{kind=link}
1. Non-Alcoholic Fatty Liver Disease and Derived Hepatocellular Carcinoma
2. Sphingolipid Metabolism: Ceramide as Central Molecule
3. Sphingolipid Contribution to Metabolic Syndrome
4. Ceramide and Other Sphingolipids in Steatosis and NASH
5. Ceramide and S1P Role in Fibrosis and Cirrhosis Development
6. Hepatocellular Carcinoma and Ceramide Metabolism
6.1. Cancer Development Implies a Reduction of Ceramide Content
6.2. DDR and S1P as Potential Targets for HCC Therapies
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellentani, S.; Scaglioni, F.; Marino, M.; Bedogni, G. Epidemiology of non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Day, C.; James, O. Steatohepatitis: A tale of two “Hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Sanyal, A.J. Mechanisms of Disease: pathogenesis of nonalcoholic fatty liver disease. Nat. Clin. Pract. Gastroenterol. Hepatol. 2005, 2, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P. From fat to inflammation. Gastroenterology 2006, 130, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Grammatikos, G.; Ferreiros, N.; Waidmann, O.; Bon, D.; Schroeter, S.; Koch, A.; Herrmann, E.; Zeuzem, S.; Kronenberger, B.; Pfeilschifter, J. Serum sphingolipid variations associate with hepatic decompensation and survival in patients with cirrhosis. PLoS ONE 2015, 10, 1–15. [Google Scholar] [CrossRef]
- Kasumov, T.; Li, L.; Li, M.; Gulshan, K.; Kirwan, J.P.; Liu, X.; Previs, S.; Willard, B.; Smith, J.D.; McCullough, A. Ceramide as a mediator of non-alcoholic fatty liver disease and associated atherosclerosis. PLoS ONE 2015, 10, 1–26. [Google Scholar] [CrossRef]
- Nagahashi, M.; Matsuda, Y.; Moro, K.; Tsuchida, J.; Soma, D.; Hirose, Y.; Kobayashi, T.; Kosugi, S.I.; Takabe, K.; Komatsu, M.; et al. DNA damage response and sphingolipid signaling in liver diseases. Surg. Today 2016, 46, 995–1005. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.; Younossi, Z.M. Epidemiology and Natural History of Non-alcoholic Fatty Liver Disease. J. Clin. Exp. Hepatol. 2012, 2, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, 99–112. [Google Scholar] [CrossRef]
- Harrison, S.A.; Torgerson, S.; Hayashi, P.H. The natural history of nonalcoholic fatty liver disease: a clinical histopathological study. Am. J. Gastroenterol. 2003, 98, 2042. [Google Scholar] [CrossRef] [PubMed]
- Samonakis, D.N.; Koulentaki, M.; Coucoutsi, C.; Augoustaki, A.; Baritaki, C.; Digenakis, E.; Papiamonis, N.; Fragaki, M.; Matrella, E.; Tzardi, M.; et al. Clinical outcomes of compensated and decompensated cirrhosis: A long term study. World J. Hepatol. 2014, 6, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Zubiete-Franco, I.; Fernandez-Tussy, P.; Barbier-Torres, L.; Simon, J.; Fernandez-Ramos, D.; Lopitz-Otsoa, F.; Gutierrez-de Juan, V.; de Davalillo, S.L.; Duce, A.M.; Iruzubieta, P.; et al. Deregulated neddylation in liver fibrosis. Hepatology 2017, 65, 694–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govaere, O.; Roskams, T. Pathogenesis and prognosis of hepatocellular carcinoma at the cellular and molecular levels. Clin. Liver Dis. 2015, 19, 261–276. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Ferlay, J. Global Cancer Statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- McGlynn, K.A.; London, W.T. The global epidemiology of hepatocellular carcinoma: present and future. Clin. Liver Dis. 2011, 15, 223–243. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma: consider the population. J. Clin. Gastroenterol. 2013, 47, S2–S6. [Google Scholar] [CrossRef] [Green Version]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Gangoiti, P.; Camacho, L.; Arana, L.; Ouro, A.; Granado, M.H.; Brizuela, L.; Casas, J.; Fabriás, G.; Abad, J.L.; Delgado, A.; et al. Control of metabolism and signaling of simple bioactive sphingolipids: Implications in disease. Prog. Lipid Res. 2010, 49, 316–334. [Google Scholar] [CrossRef]
- Gomez-Munoz, A. Ceramide 1-phosphate/ceramide, a switch between life and death. Biochim. Biophys. Acta 2006, 1758, 2049–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haimovitz-Friedman, A.; Kolesnick, R.N.; Fuks, Z. Ceramide signaling in apoptosis. Br. Med. Bull. 1997, 53, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Mullen, T.D.; Obeid, L.M. Ceramide and apoptosis: exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer Agents Med. Chem. 2012, 12, 340–363. [Google Scholar] [CrossRef] [PubMed]
- Albeituni, S.; Stiban, J. Roles of Ceramides and Other Sphingolipids in Immune Cell Function and Inflammation. Adv. Exp. Med. Biol. 2019, 1161, 169–191. [Google Scholar] [PubMed]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2012, in press. [Google Scholar] [CrossRef]
- Arana, L.; Ordonez, M.; Ouro, A.; Rivera, I.-G.; Gangoiti, P.; Trueba, M.; Gomez-Munoz, A. Ceramide 1-phosphate induces macrophage chemoattractant protein-1 release: involvement in ceramide 1-phosphate-stimulated cell migration. AJP Endocrinol. Metab. 2013, 304, E1213–E1226. [Google Scholar] [CrossRef]
- Granado, M.H.; Gangoiti, P.; Ouro, A.; Arana, L.; González, M.; Trueba, M.; Gómez-Muñoz, A. Ceramide 1-phosphate (C1P) promotes cell migration. Involvement of a specific C1P receptor. Cell. Signal. 2009, 21, 405–412. [Google Scholar] [CrossRef]
- Ouro, A.; Arana, L.; Rivera, I.G.; Ordoñez, M.; Gomez-Larrauri, A.; Presa, N.; Simón, J.; Trueba, M.; Gangoiti, P.; Bittman, R.; et al. Phosphatidic acid inhibits ceramide 1-phosphate-stimulated macrophage migration. Biochem. Pharmacol. 2014, 92, 642–650. [Google Scholar] [CrossRef]
- Lee, M.J.; Van Brocklyn, J.R.; Thangada, S.; Liu, C.H.; Hand, A.R.; Menzeleev, R.; Spiegel, S.; Hla, T. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science 1998, 279, 1552–1555. [Google Scholar] [CrossRef]
- Taha, T.A.; Hannun, Y.A.; Obeid, L.M. Sphingosine kinase: biochemical and cellular regulation and role in disease. J. Biochem. Mol. Biol. 2006, 39, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Mandala, S.M. Sphingosine-1-phosphate phosphatases. Prostaglandins Other Lipid Mediat. 2001, 64, 143–156. [Google Scholar] [CrossRef]
- Johnson, K.R.; Johnson, K.Y.; Becker, K.P.; Bielawski, J.; Mao, C.; Obeid, L.M. Role of human sphingosine-1-phosphate phosphatase 1 in the regulation of intra- and extracellular sphingosine-1-phosphate levels and cell viability. J. Biol. Chem. 2003, 278, 34541–34547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestri, A.; Pierucci, F.; Frati, A.; Monaco, L.; Meacci, E. Sphingosine 1-Phosphate Receptors: Do They Have a Therapeutic Potential in Cardiac Fibrosis? Front. Pharmacol. 2017, 8, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetzl, E.J.; Wang, W.; McGiffert, C.; Huang, M.C.; Graler, M.H. Sphingosine 1-phosphate and its G protein-coupled receptors constitute a multifunctional immunoregulatory system. J. Cell. Biochem. 2004, 92, 1104–1114. [Google Scholar] [CrossRef] [Green Version]
- Grammatikos, G.; Mühle, C.; Ferreiros, N.; Schroeter, S.; Bogdanou, D.; Schwalm, S.; Hintereder, G.; Kornhuber, J.; Zeuzem, S.; Sarrazin, C.; et al. Serum acid sphingomyelinase is upregulated in chronic hepatitis C infection and non alcoholic fatty liver disease. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 2014, 1841, 1012–1020. [Google Scholar] [CrossRef]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Brönneke, H.S.; et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef] [Green Version]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of Ceramide Synthesis Ameliorates Glucocorticoid-, Saturated-Fat-, and Obesity-Induced Insulin Resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Cinar, R.; Godlewski, G.; Liu, J.; Tam, J.; Jourdan, T.; Mukhopadhyay, B.; Harvey-White, J.; Kunos, G. Hepatic cannabinoid-1 receptors mediate diet-induced insulin resistance by increasing de novo synthesis of long-chain ceramides. Hepatology 2014, 59, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikman, B.T.; Guan, Y.; Shui, G.; Siddique, M.M.; Holland, W.L.; Kim, J.Y.; Fabriàs, G.; Wenk, M.R.; Summers, S.A. Fenretinide prevents lipid-induced insulin resistance by blocking ceramide biosynthesis. J. Biol. Chem. 2012, 287, 17426–17437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, M.L.; Coghlan, M.; Hundal, H.S. Modulating serine palmitoyl transferase (SPT) expression and activity unveils a crucial role in lipid-induced insulin resistance in rat skeletal muscle cells. Biochem. J. 2009, 417, 791–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, L.; Junlin, Z.; Aimin, W.; Niansheng, L.; Benmei, C.; Minxiang, L. Inhibition of ceramide synthesis reverses endothelial dysfunction and atherosclerosis in streptozotocin-induced diabetic rats. Diabetes Res. Clin. Pract. 2011, 93, 77–85. [Google Scholar] [CrossRef]
- Glaros, E.N.; Kim, W.S.; Wu, B.J.; Suarna, C.; Quinn, C.M.; Rye, K.A.; Stocker, R.; Jessup, W.; Garner, B. Inhibition of atherosclerosis by the serine palmitoyl transferase inhibitor myriocin is associated with reduced plasma glycosphingolipid concentration. Biochem. Pharmacol. 2007, 73, 1340–1346. [Google Scholar] [CrossRef]
- Yang, G.; Badeanlou, L.; Bielawski, J.; Roberts, A.J.; Hannun, Y.A.; Samad, F. Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2009, 297. [Google Scholar] [CrossRef] [Green Version]
- Park, T.S.; Panek, R.L.; Rekhter, M.D.; Mueller, S.B.; Rosebury, W.S.; Robertson, A.; Hanselman, J.C.; Kindt, E.; Homan, R.; Karathanasis, S.K. Modulation of lipoprotein metabolism by inhibition of sphingomyelin synthesis in ApoE knockout mice. Atherosclerosis 2006, 189, 264–272. [Google Scholar] [CrossRef]
- Zabielski, P.; Daniluk, J.; Hady, H.R.; Markowski, A.R.; Imierska, M.; Gorski, J.; Blachnio-Zabielska, A.U. The effect of high-fat diet and inhibition of ceramide production on insulin action in liver. J. Cell. Physiol. 2019, 234, 1851–1861. [Google Scholar] [CrossRef]
- Samad, F.; Badeanlou, L.; Shah, C.; Yang, G. Adipose tissue and ceramide biosynthesis in the pathogenesis of obesity. Adv. Exp. Med. Biol. 2011, 721, 67–86. [Google Scholar]
- Chaurasia, B.; Summers, S.A. Ceramides - Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol. Metab. 2015, 26, 538–550. [Google Scholar] [CrossRef]
- Chaurasia, B.; Kaddai, V.A.; Lancaster, G.I.; Henstridge, D.C.; Sriram, S.; Galam, D.L.A.; Gopalan, V.; Prakash, K.N.B.; Velan, S.S.; Bulchand, S.; et al. Adipocyte Ceramides Regulate Subcutaneous Adipose Browning, Inflammation, and Metabolism. Cell Metab. 2016, 24, 820–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- JeBailey, L.; Wanono, O.; Niu, W.; Roessler, J.; Rudich, A.; Klip, A. Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes 2007, 56, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribaux, P.G.; Iynedjian, P.B. Analysis of the role of protein kinase B (cAKT) in insulin-dependent induction of glucokinase and sterol regulatory element-binding protein 1 (SREBP1) mRNAs in hepatocytes. Biochem. J. 2003, 376, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Summers, S.A. Sphingolipids, insulin resistance, and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 2008, 29, 381–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summers, S.A. Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 2006, 45, 42–72. [Google Scholar] [CrossRef] [PubMed]
- Zubiete-Franco, I.; Garcia-Rodriguez, J.L.; Martinez-Una, M.; Martinez-Lopez, N.; Woodhoo, A.; Juan, V.G.-D.; Beraza, N.; Lage-Medina, S.; Andrade, F.; Fernandez, M.L.; et al. Methionine and S-adenosylmethionine levels are critical regulators of PP2A activity modulating lipophagy during steatosis. J. Hepatol. 2016, 64, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, G.M.; Carey, A.L.; Selathurai, A.; Kingwell, B.A.; Bruce, C.R. Plasma Sphingosine-1-Phosphate Is Elevated in Obesity. PLoS ONE 2013, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Kowalski, G.M.; Leslie, S.J.; Risis, S.; Yang, C.; Lee-Young, R.S.; Babb, J.R.; Meikle, P.J.; Lancaster, G.I.; Henstridge, D.C.; et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 2013, 56, 1638–1648. [Google Scholar] [CrossRef]
- Fayyaz, S.; Henkel, J.; Japtok, L.; Krämer, S.; Damm, G.; Seehofer, D.; Püschel, G.P.; Kleuser, B. Involvement of sphingosine 1-phosphate in palmitate-induced insulin resistance of hepatocytes via the S1P2 receptor subtype. Diabetologia 2014, 57, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty acid-induced β cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Bielawski, J.; Samad, F.; Merrill, A.H.; Cowart, L.A. Palmitate increases sphingosine-1-phosphate in C2C12 myotubes via upregulation of sphingosine kinase message and activity. J. Lipid Res. 2009, 50, 1852–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasumov, T.; Huang, H.; Chung, Y.M.; Zhang, R.; McCullough, A.J.; Kirwan, J.P. Quantification of ceramide species in biological samples by liquid chromatography electrospray ionization tandem mass spectrometry. Anal. Biochem. 2010, 401, 154–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Kotani, K.; Tanaka, K.; Egucih, Y.; Anzai, K. Therapeutic approaches to nonalcoholic fatty liver disease: Exercise intervention and related mechanisms. Front. Endocrinol. (Lausanne) 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.; Kennedy, L.; Hargrove, L.; Demieville, J.; Thomson, J.; Alpini, G.; Francis, H. Updates on dietary models of nonalcoholic fatty liver disease: Current studies and insights. Gene Expr. 2018, 18, 5–17. [Google Scholar] [CrossRef]
- Bektas, M.; Laura Allende, M.; Lee, B.G.; Chen, W.P.; Amar, M.J.; Remaley, A.T.; Saba, J.D.; Proia, R.L. Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J. Biol. Chem. 2010, 285, 10880–10889. [Google Scholar] [CrossRef] [Green Version]
- Aguilera-Romero, A.; Gehin, C.; Riezman, H. Sphingolipid homeostasis in the web of metabolic routes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 647–656. [Google Scholar] [CrossRef]
- Kotronen, A.; Seppänen-Laakso, T.; Westerbacka, J.; Kiviluoto, T.; Arola, J.; Ruskeepää, A.L.; Yki-Järvinen, H.; Orešič, M. Comparison of lipid and fatty acid composition of the liver, subcutaneous and intra-abdominal adipose tissue, and serum. Obesity 2010, 18, 937–944. [Google Scholar] [CrossRef]
- Llacuna, L.; Marí, M.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Morales, A. Critical role of acidic sphingomyelinase in murine hepatic ischemia-reperfusion injury. Hepatology 2006, 44, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Chocian, G.; Chabowski, A.; Żendzian-Piotrowska, M.; Harasim, E.; Łukaszuk, B.; Górski, J. High fat diet induces ceramide and sphingomyelin formation in rat’s liver nuclei. Mol. Cell. Biochem. 2010, 340, 125–131. [Google Scholar] [CrossRef]
- Cano, A.; Buqué, X.; Martínez-uña, M.; Aurrekoetxea, I.; Menor, A.; García-rodriguez, J.L.; Lu, S.C.; Martínez-chantar, M.L.; Mato, J.M. Methionine adenosyltransferase 1A gene deletion disrupts hepatic VLDL assembly in mice. Hepatology 2011, 54, 1975–1986. [Google Scholar] [CrossRef] [Green Version]
- Presa, N.; Clugston, R.D.; Lingrell, S.; Kelly, S.E.; Merrill, A.H.; Jana, S.; Kassiri, Z.; Gómez-Muñoz, A.; Vance, D.E.; Jacobs, R.L.; et al. Vitamin E alleviates non-alcoholic fatty liver disease in phosphatidylethanolamine N-methyltransferase deficient mice. Biochim. Biophys. Acta - Mol. Basis Dis. 2019, 1865, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Fernández-Ramos, D.; Varela-Rey, M.; Martínez-Arranz, I.; Navasa, N.; Van Liempd, S.M.; Lavín Trueba, J.L.; Mayo, R.; Ilisso, C.P.; de Juan, V.G.; et al. Metabolomic Identification of Subtypes of Nonalcoholic Steatohepatitis. Gastroenterology 2017, 152, 1449–1461.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monetti, M.; Levin, M.C.; Watt, M.J.; Sajan, M.P.; Marmor, S.; Hubbard, B.K.; Stevens, R.D.D.; Bain, J.R.; Newgard, C.B.; Farese, R.V.; et al. Dissociation of Hepatic Steatosis and Insulin Resistance in Mice Overexpressing DGAT in the Liver. Cell Metab. 2007, 6, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goni, F.M.; Alonso, A. Sphingomyelinases: enzymology and membrane activity. FEBS Lett. 2002, 531, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Deevska, G.M.; Rozenova, K.A.; Giltiay, N.V.; Chambers, M.A.; White, J.; Boyanovsky, B.B.; Wei, J.; Daugherty, A.; Smart, E.J.; Reid, M.B.; et al. Acid sphingomyelinase deficiency prevents diet-induced hepatic triacylglycerol accumulation and hyperglycemia in mice. J. Biol. Chem. 2009, 284, 8359–8368. [Google Scholar] [CrossRef] [Green Version]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef]
- Gorden, D.L.; Myers, D.S.; Ivanova, P.T.; Fahy, E.; Maurya, M.R.; Gupta, S.; Min, J.; Spann, N.J.; McDonald, J.G.; Kelly, S.L.; et al. Biomarkers of NAFLD progression: A lipidomics approach to an epidemic 1. J. Lipid Res. 2015, 56, 722–736. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Fernández-Checa, J.C. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species: Role of mitochondrial glutathione. J. Biol. Chem. 1997, 272, 11369–11377. [Google Scholar] [CrossRef] [Green Version]
- Raichur, S.; Wang, S.T.; Chan, P.W.; Li, Y.; Ching, J.; Chaurasia, B.; Dogra, S.; Öhman, M.K.; Takeda, K.; Sugii, S.; et al. CerS2 haploinsufficiency inhibits β-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab. 2014, 20, 687–695. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, H.; Gores, G. Mechanisms of Liver Injury: An Overview. Curr. Mol. Med. 2003, 3, 483–490. [Google Scholar] [CrossRef]
- Chang, Z.Q.; Lee, S.Y.; Kim, H.J.; Kim, J.R.; Kim, S.J.; Hong, I.K.; Oh, B.C.; Choi, C.S.; Goldberg, I.J.; Park, T.S. Endotoxin activates de novo sphingolipid biosynthesis via nuclear factor kappa B-mediated upregulation of Sptlc2. Prostaglandins Other Lipid Mediat. 2011, 94, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolova-Karakashian, M.N.; Rozenova, K.A. Ceramide in stress response. Adv. Exp. Med. Biol. 2010, 688, 86–108. [Google Scholar] [PubMed]
- Marí, M.; Fernández-Checa, J.C. Sphingolipid signalling and liver diseases. Liver Int. 2007, 27, 440–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, R.F.; Brenner, D.A. Mechanisms of liver injury. I. TNF-α-induced liver injury: Role of IKK, JNK, and ROS pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, 583–589. [Google Scholar] [CrossRef]
- Clarke, C.J.; Snook, C.F.; Tani, M.; Matmati, N.; Marchesini, N.; Hannun, Y.A. The extended family of neutral sphingomyelinases. Biochemistry 2006, 45, 11247–11256. [Google Scholar] [CrossRef]
- Nikolova-Karakashian, M.; Karakashian, A.; Rutkute, K. Role of neutral sphingomyelinases in aging and inflammation. Subcell. Biochem. 2008, 49, 469–486. [Google Scholar]
- Polyzos, S.A.; Toulis, K.A.; Goulis, D.G.; Zavos, C.; Kountouras, J. Serum total adiponectin in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Metabolism 2011, 60, 313–326. [Google Scholar] [CrossRef]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide Is Metabolized to Acylceramide and Stored in Lipid Droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef] [Green Version]
- Tsugane, K.; Tamiya-Koizumi, K.; Nagino, M.; Nimura, Y.; Yoshida, S. A possible role of nuclear ceramide and sphingosine in hepatocyte apoptosis in rat liver. J. Hepatol. 1999, 31, 8–17. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- NICE Guideline [NG49]. Non-alcoholic Fatty Liver Disease: Assessment and Management. 2016, Volume 34. Available online: https://www.nice.org.uk/guidance/ng49 (accessed on 9 October 2019).
- Pessayre, D.; Mansouri, A.; Fromenty, B.; Mansouri, A. Nonalcoholic Steatosis and Steatohepatitis V. Mitochondrial dysfunction in steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, 193–199. [Google Scholar] [CrossRef]
- Schuppan, D.; Afdhal, N.H. Liver Cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar]
- Shea, B.S.; Tager, A.M. Sphingolipid Regulation of Tissue Fibrosis. Open Rheumatol. J. 2012, 6, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Guicciardi, M.E.; Gores, G.J. Hepatocyte death: a clear and present danger. Physiol. Rev. 2010, 90, 1165–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Ramos, D.; Fernandez-Tussy, P.; Lopitz-Otsoa, F.; Gutierrez-de-Juan, V.; Navasa, N.; Barbier-Torres, L.; Zubiete-Franco, I.; Simon, J.; Fernandez, A.F.; Arbelaiz, A.; et al. MiR-873-5p acts as an epigenetic regulator in early stages of liver fibrosis and cirrhosis. Cell Death Dis. 2018, 9, 958. [Google Scholar] [CrossRef]
- Ichi, I.; Nakahara, K.; Fujii, K.; Iida, C.; Miyashita, Y.; Kojo, S. Increase of ceramide in the liver and plasma after carbon tetrachloride intoxication in the rat. J. Nutr. Sci. Vitaminol. (Tokyo) 2007, 53, 53–56. [Google Scholar] [CrossRef] [Green Version]
- Mari, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef]
- Mari, M.; Colell, A.; Morales, A.; Caballero, F.; Moles, A.; Fernandez, A.; Terrones, O.; Basanez, G.; Antonsson, B.; Garcia-Ruiz, C.; et al. Mechanism of mitochondrial glutathione-dependent hepatocellular susceptibility to TNF despite NF-kappaB activation. Gastroenterology 2008, 134, 1507–1520. [Google Scholar] [CrossRef]
- Moles, A.; Tarrats, N.; Morales, A.; Domínguez, M.; Bataller, R.; Caballería, J.; García-Ruiz, C.; Fernández-Checa, J.C.; Marí, M. Acidic sphingomyelinase controls hepatic stellate cell activation and in vivo liver fibrogenesis. Am. J. Pathol. 2010, 177, 1214–1224. [Google Scholar] [CrossRef] [Green Version]
- Changyong, L.; Xiangming, J.; Lin, Y.; Xihong, L.; Shi, Y.; Liying, L. Involvement of sphingosine 1-phosphate (SIP)/S1P3 signaling in cholestasis-induced liver fibrosis. Am. J. Pathol. 2009, 175, 1464–1472. [Google Scholar]
- Li, C.; Zheng, S.; You, H.; Liu, X.; Lin, M.; Yang, L.; Li, L. Sphingosine 1-phosphate (S1P)/S1P receptors are involved in human liver fibrosis by action on hepatic myofibroblasts motility. J. Hepatol. 2011, 54, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yue, S.; Li, C.; Yang, L.; You, H.; Li, L. Essential roles of sphingosine 1-phosphate receptor types 1 and 3 in human hepatic stellate cells motility and activation. J. Cell. Physiol. 2011, 226, 2370–2377. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Watanabe, N.; Ishii, I.; Shimosawa, T.; Kume, Y.; Tomiya, T.; Inoue, Y.; Nishikawa, T.; Ohtomo, N.; Tanoue, Y.; et al. Sphingosine 1-phosphate regulates regeneration and fibrosis after liver injury via sphingosine 1-phosphate receptor 2. J. Lipid Res. 2009, 50, 556–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davaille, J.; Li, L.; Mallat, A.; Lotersztajn, S. Sphingosine 1-phosphate triggers both apoptotic and survival signals for human hepatic myofibroblasts. J. Biol. Chem. 2002, 277, 37323–37330. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Goodman, Z.; Sanyal, A. Review Current efforts and trends in the treatment of NASH. J. Hepatol. 2015, 62, S65–S75. [Google Scholar] [CrossRef] [Green Version]
- Benyon, R.; Iredale, J. Is liver fibrosis reversible? Gut 2000, 46, 443–446. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Li, Z.; Li, H.; Li, L.; Li, J.; Yu, C. Metabolic profile changes of CCl4-liver fibrosis and inhibitory effects of Jiaqi Ganxian granule. Molecules 2016, 21, 698. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Meng, H.Y.; Liu, S.M.; Wang, Y.; Yang, X.X.; Lu, F.; Wang, H.Y. Identification of key metabolic changes during liver fibrosis progression in rats using a urine and serum metabolomics approach. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mücke, V.T.; Thomas, D.; Mücke, M.M.; Waidmann, O.; Zeuzem, S.; Sarrazin, C.; Pfeilschifter, J.; Vermehren, J.; Finkelmeier, F.; Grammatikos, G. Serum sphingolipids predict de novo hepatocellular carcinoma in hepatitis C cirrhotic patients with sustained virologic response. Liver Int. 2019, 1–10. [Google Scholar] [CrossRef]
- Krautbauer, S.; Wiest, R.; Liebisch, G.; Buechler, C. Associations of systemic sphingolipids with measures of hepatic function in liver cirrhosis are related to cholesterol. Prostaglandins Other Lipid Mediat. 2017, 131, 25–32. [Google Scholar] [CrossRef]
- Duan, Z.P. Plasma sphingolipids as potential indicators of hepatic necroinflammation in patients with chronic hepatitis C and normal alanine aminotransferase level. PLoS ONE 2014, 9, e95095. [Google Scholar]
- Attwa, M.H.; El-Etreby, S.A. Guide for diagnosis and treatment of hepatocellular carcinoma. World J. Hepatol. 2015, 7, 1632–1651. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Marrero, J.A.; Rudolph, L.; Reddy, K.R. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology 2008, 134, 1752–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Li, Z.; Guan, M.; Lin, Y.; Cui, X.; Zhang, Y.; Zhao, Z.; Zhu, J. Aberrant lipid metabolism in hepatocellular carcinoma revealed by liver lipidomics. Int. J. Mol. Sci. 2017, 18, 2550. [Google Scholar] [CrossRef] [Green Version]
- Takashima, Y.; Horisawa, K.; Udono, M.; Ohkawa, Y.; Suzuki, A. Prolonged inhibition of hepatocellular carcinoma cell proliferation by combinatorial expression of defined transcription factors. Cancer Sci. 2018, 109, 3543–3553. [Google Scholar] [CrossRef]
- Hajj, C.; Becker-flegler, K.A.; Haimovitz-friedman, A. Novel mechanisms of action of classical chemotherapeutic agents on sphingolipid pathways. Biol. Chem. 2015, 396, 669–679. [Google Scholar] [CrossRef]
- Bonnaud, S.; Niaudet, C.; Legoux, F.; Corre, I.; Delpon, G.; Saulquin, X. Sphingosine-1-Phosphate Activates the AKT Pathway to Protect Small Intestines from Radiation-Induced Endothelial Apoptosis. Ther. Targets Chem. Biol. 2010, 70, 9905–9915. [Google Scholar] [CrossRef] [Green Version]
- Carroll, B.; Donaldson, J.C.; Obeid, L. Sphingolipids in the DNA damage response. Adv. Biol. Regul. 2015, 58, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yu-Teh, L. Ceramide Glycosilation Catalyzed by Glucosylceramide Synthase and Cancer Drug Resistance. Adv. Cancer Res. 2013, 117, 59–89. [Google Scholar]
- Gómez-muñoz, A.; Kong, J.Y.; Salh, B.; Steinbrecher, U.P.; Duffy, P.A.; Martin, A.; Brien, L.O.; Byun, H.; Pharmacol, D.N.B.M.; Frago, L.M.; et al. Ceramide-1-phosphate blocks apoptosis through inhibition of acid sphingomyelinase in macrophages. J. Lipid Res. 2004, 45, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangoiti, P.; Granado, M.H.; Wei, S.; Kong, J.Y.; Steinbrecher, U.P.; Gómez-muñoz, A. Ceramide 1-phosphate stimulates macrophage proliferation through activation of the PI3-kinase/PKB, JNK and ERK1/2 pathways. Cell. Signal. 2008, 20, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Kolesnick, R.; Altieri, D.; Fuks, Z. A CERTain Role for Ceramide. Cancer Cell 2007, 473–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanton, C.; Marani, M.; Pardo, O.; Warne, P.H.; Kelly, G.; Sahai, E.; Chang, J.; Temple, J.; Ahmed, A.A.; Brenton, J.D.; et al. Article Regulators of Mitotic Arrest and Ceramide Metabolism Are Determinants of Sensitivity to Paclitaxel and Other Chemotherapeutic Drugs. Cancer Cell 2007, 11, 498–512. [Google Scholar] [CrossRef] [Green Version]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Bose, R.; Verheij, M.; Haimovitz-friedman, A.; Scotto, K.; Fuks, Z.; Kolesnick, R. Ceramide Synthase Mediates Daunorubicin-lnduced Apoptosis: An Alternative Mechanism for Generating Death Signals. Cell 1995, 82, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Dumitru, C.A.; Sandalcioglu, I.E.; Wagner, M.; Weller, M.; Gulbins, E. Lysosomal ceramide mediates gemcitabine-induced death of glioma cells. J. Mol. Med. 2009, 87, 1123–1132. [Google Scholar] [CrossRef] [Green Version]
- Lacour, S.; Hammann, A.; Grazide, S.; Lagadic-Gossmann, D.; Athias, A.; Sergent, O.; Laurent, G.; Gambert, P.; Solary, E.; Dimanche-Boitrel, M.-T. Cisplatin-induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Cancer Res. 2004, 64, 3593–3598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.; Stegner, A.L.; Alexander, H.; Alexander, S. Overexpression of Sphingosine-1-Phosphate Lyase or Inhibition of Sphingosine Kinase in Dictyostelium discoideum Results in a Selective Increase in Sensitivity to Platinum-Based Chemotherapy Drugs. Eukaryot. Cell 2004, 3, 795–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.; Mesika, A.; Sivaguru, M.; Van Veldhoven, P.P.; Alexander, H.; Futerman, A.H.; Alexander, S. (Dihydro)ceramide Synthase 1 – Regulated Sensitivity to Cisplatin Is Associated with the Activation of p38 Mitogen-Activated Protein Kinase and Is Abrogated by Sphingosine Kinase 1. Mol. Cancer Res. 2007, 5, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Cabot, M.C.; Han, T.; Giuliano, A.E. The multidrug resistance modulator SDZ PSC 833 is a potent activator of cellular ceramide formation. FEBS Lett. 1998, 431, 185–188. [Google Scholar] [CrossRef] [Green Version]
- Xie, R.; Nguyen, S.; Mckeehan, W.L.; Liu, L. Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol. 2010, 11, 89. [Google Scholar] [CrossRef] [Green Version]
- Savic, R.; Xingxuan, H.; Fiel, I.; Schuchman, E.H. Recombinant Human Acid Sphingomyelinase as an Adjuvant to Sorafenib Treatment of Experimental Liver Cancer. PLoS ONE 2013, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berasain, C. Hepatocellular carcinoma and sorafenib: too many resistance mechanisms? Gut 2013, 62, 1674–1675. [Google Scholar] [CrossRef] [PubMed]
- Freeman, B.A.; Crapo, J.D. Biology of disease: free radicals and tissue injury. Lab. Investig. 1982, 47, 412–426. [Google Scholar]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, S.; Kleuser, B.; Scha, M.; Kim, K.; Park, K. Sphingosine-1-phosphate inhibits human keratinocyte proliferation via Akt / protein kinase B inactivation. Cell. Signal. 2004, 16, 89–95. [Google Scholar] [CrossRef]
- Matsuda, Y.; Wakai, T.; Hirose, Y.; Osawa, M.; Fujimaki, S. p27 Is a Critical Prognostic Biomarker in Non-Alcoholic Steatohepatitis-Related Hepatocellular Carcinoma. Int. J. Mol. Sci. 2013, 14, 23499–23515. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Xu, M.; Gao, J.; Li, M. Alkaline ceramidase 3 promotes growth of hepatocellular carcinoma cells via regulating S1P/S1PR2/PI3K/AKT signaling. Pathol. Res. Pr. 2018, 214, 1381–1387. [Google Scholar] [CrossRef]
- Osawa, Y.; Nagaki, M.; Banno, Y.; Nozawa, Y.; Moriwaki, H.; Nakashima, S. Sphingosine Kinase Regulates Hepatoma Cell Differentiation: Roles of Hepatocyte Nuclear Factor and Retinoid Receptor. Biochem. Biophys. Res. Commun. 2001, 286, 673–677. [Google Scholar] [CrossRef]
- Martínez-chantar, M.L.; García-Trevijano, E.R.; Latasa, M.U.; Martín-Duce, A.; Fortes, P.; Caballería, J.; Ávila, M.A.; Mato, J.M. Methionine Adenosyltransferase II Beta Subunit Gene Expression Provides a Proliferative Advantage in Human Hepatoma. Gastroenterology 2003, 124, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Zubiete-franco, I.; García-rodríguez, J.L.; Lopitz-otsoa, F.; Serrano-macia, M.; Simon, J.; Fernández-tussy, P.; Barbier-torres, L.; Fernández-ramos, D.; Gutiérrez-de-juan, V.; López, S.; et al. SUMOylation regulates LKB1 localization and its oncogenic activity in liver cancer. EBioMedicine 2019, 40, 406–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruckhäveberle, E.; Rody, A.; Engels, K.; Gaetje, R.; von Minckwitz, G.; Schiffman, S.; Grösch, S.; Geisslinger, G.; Holtrich, U.; Karn, T.; et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res. Treat. 2008, 41–52. [Google Scholar]
- Liu, S.; Su, Y.; Qin, M.; Mao, Y.; Huang, J.; Tang, G. Sphingosine kinase 1 promotes tumor progression and confers malignancy phenotypes of colon cancer by regulating the focal adhesion kinase pathway and adhesion molecules. Int. J. Oncol. 2013, 42, 617–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahashi, M.; Ramachandra, S.; Kim, E.Y.; Allegood, J.C.; Rashid, O.M.; Yamada, A.; Zhao, R.; Milstien, S.; Zhou, H.; Spiegel, S.; et al. Sphingosine 1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012, 72, 726–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Barbier-torres, L.; Beraza, N.; Fernández-tussy, P.; Lopitz-, F.; Fernández-ramos, D.; Zubiete-franco, I.; Varela-rey, M.; Teresa, C.; Gutiérrez, V.; Anguita, J.; et al. Histone Deacetylase 4 promotes cholectatic liver injury in the absence of Prohibitin-1. Hepatology 2015, 62, 1237–1248. [Google Scholar] [CrossRef] [Green Version]
- Barbier-torres, L.; Delgado, T.C.; García-rodríguez, J.L.; Fernández-ramos, D.; Buqué, X.; Cano, A.; Boix, L.; Bruix, J.; Villa, E.; Castro, A.; et al. Stabilization of LKB1 and Akt by neddylation regulates energy metabolism in liver cancer. Oncotarget 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Critelli, R.; Milosa, F.; Faillaci, F.; Condello, R.; Turola, E.; Marzi, L.; Lei, B.; Dituri, F.; Andreani, S.; Sighinolfi, P.; et al. Microenvironment inflammatory infiltrate drives growth speed and outcome of hepatocellular carcinoma: a prospective clinical study. Cell Death Dis. 2017, 8, e3017. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, J.; Ouro, A.; Ala-Ibanibo, L.; Presa, N.; Delgado, T.C.; Martínez-Chantar, M.L. Sphingolipids in Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma: Ceramide Turnover. Int. J. Mol. Sci. 2020, 21, 40. https://doi.org/10.3390/ijms21010040
Simon J, Ouro A, Ala-Ibanibo L, Presa N, Delgado TC, Martínez-Chantar ML. Sphingolipids in Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma: Ceramide Turnover. International Journal of Molecular Sciences. 2020; 21(1):40. https://doi.org/10.3390/ijms21010040
Chicago/Turabian StyleSimon, Jorge, Alberto Ouro, Lolia Ala-Ibanibo, Natalia Presa, Teresa Cardoso Delgado, and María Luz Martínez-Chantar. 2020. "Sphingolipids in Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma: Ceramide Turnover" International Journal of Molecular Sciences 21, no. 1: 40. https://doi.org/10.3390/ijms21010040
APA StyleSimon, J., Ouro, A., Ala-Ibanibo, L., Presa, N., Delgado, T. C., & Martínez-Chantar, M. L. (2020). Sphingolipids in Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma: Ceramide Turnover. International Journal of Molecular Sciences, 21(1), 40. https://doi.org/10.3390/ijms21010040