Human Molecular Chaperone Hsp60 and Its Apical Domain Suppress Amyloid Fibril Formation of α-Synuclein


 and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Open-Mimic Mutant Hsp60 G190W (GW) Is a Potent Suppressor of Amyloid Fibril Formation
2.2. Hsp60 GW Shows an Increased Degree of Surface Hydrophobicity Due to Open-Mimic AD Conformation
2.3. Hsp60 WT and GW Interact with α-Synuclein Monomers
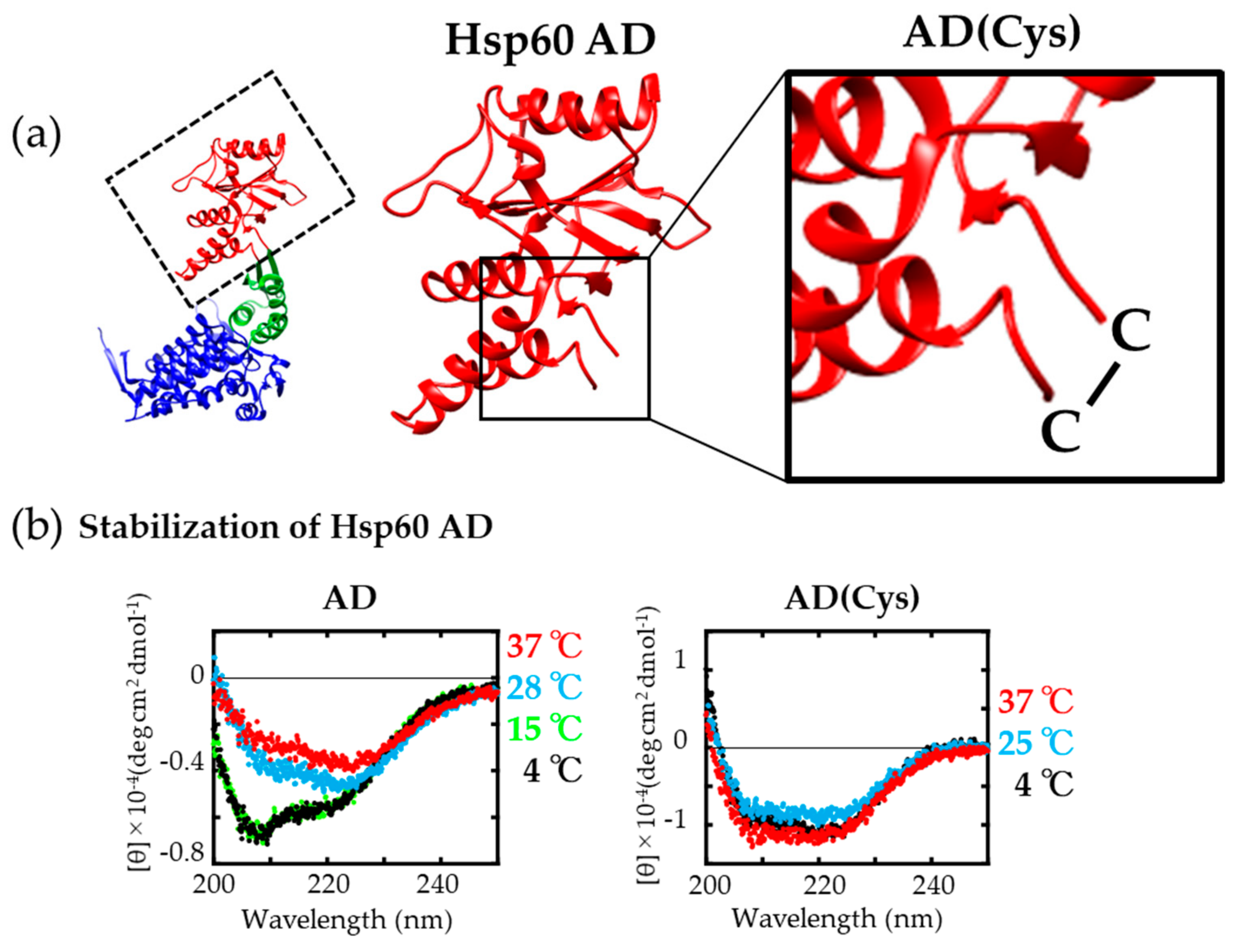
2.4. Stabilization of Isolated Hsp60 AD
2.5. Hsp60 AD Functions as a Mini-Chaperone
2.6. Hsp60 AD Suppresses α-Synuclein Aggregation and Cytotoxicity
3. Materials and Methods
3.1. Materials
3.2. Protein Expression and Purification
3.3. Thioflavin-T (ThT) Fluorescence Assays
3.4. Transmission Electron Microscopy (TEM)
3.5. Circular Dichroism (CD)
3.6. 8-Anilino-1-Naphthalenesulfonic Acid (ANS) Fluorescence Assays
3.7. Gel-Filtration Chromatography Analysis
3.8. Immunoprecipitation
3.9. Western Blot Analysis
3.10. Quartz Crystal Microbalance (QCM) Assay
3.11. Cells
3.11.1. Cell Cultures
3.11.2. Construction of pCAG-GFP-Neo Gene
3.11.3. Generation of GFP-Syn Stable Cell Lines (GFP-Syn N2a Cells)
3.12. Introduction of Hsp60AD(Cys) Protein into Cells
3.13. Measurement of Cell Death
3.14. Immunofluorescence Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Apical domain |
| ANS | Anilino-1-naphthalenesulfonic acid |
| CD | Circular dichroism |
| C2-NTA | 3,3′-Dithiobis[N-(5-amino-5-carboxypentyl) propionamide-N,N’-diacetic acid] dihydrochloride |
| EthD-1 | Ethidium Homodimer-1 |
| FBS | Fetal bovine serum |
| GFP-Syn | Green fluorescent protein-tagged human α-synuclein |
| GW | Hsp60 G190W mutant |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| MEM | Minimum Essential Medium |
| N2a | Neuro2a |
| Neo | Neomycin/kanamycin resistance gene |
| NHS | N-hydroxysuccinimide |
| 6-OHDA | 6-hydroxydopamine |
| PBS | Phosphate-Buffered Saline |
| PCR | Polymerase chain reaction |
| PD | Parkinson’s disease |
| PDB | Protein Data Bank |
| PMSF | Phenylmethylsulfonyl fluoride |
| polyQ | Polyglutamine |
| QCM | Quartz crystal microbalance |
| RFU | Relative Fluorescence Unit |
| SAM | Self-assembled monolayer |
| SEM | Standard error of the mean |
| sHsp | Small heat shock protein |
| TEM | Transmission Electron Microscopy |
| ThT | Thioflavin T |
| WSC | 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide |
| WT | Hsp60 wild type |
References
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Folding proteins in fatal ways. Nature 2003, 426, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.; Beal, M.F. Parkinson’s disease. Hum. Mol. Genet. 2007, 16, R183–R194. [Google Scholar] [CrossRef] [Green Version]
- Arosio, P.; Knowles, T.P.J.; Linse, S. On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef] [Green Version]
- Ferrone, F. Analysis of protein aggregation kinetics. Methods Enzymol. 1999, 309, 256–274. [Google Scholar]
- Lee, C.C.; Nayak, A.; Sethuraman, A.; Belfort, G.; McRae, G.J. A three-stage kinetic model of amyloid fibrillation. Biophys. J. 2007, 92, 3448–3458. [Google Scholar] [CrossRef] [Green Version]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [Green Version]
- McLean, P.J.; Kawamata, H.; Shariff, S.; Hewett, J.; Sharma, N.; Ueda, K.; Breakefield, X.O.; Hyman, B.T. TorsinA and heat shock proteins act as molecular chaperones: Suppression of alpha-synuclein aggregation. J. Neurochem. 2002, 83, 846–854. [Google Scholar] [CrossRef]
- Auluck, P.K.; Chan, H.Y.E.; Trojanowski, J.Q.; Lee, V.M.Y.; Bonini, N.M. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef]
- Diaz-Villanueva, J.F.; Diaz-Molina, R.; Garcia-Gonzalez, V. Protein Folding and Mechanisms of Proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.; Carver, J.A.; Ecroyd, H. Preventing alpha-synuclein aggregation: The role of the small heat-shock molecular chaperone proteins. Biochim. Biophys. Acta-Mol. Basis Dis. 2014, 1842, 1830–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolhassani, A.; Rafati, S. Mini-chaperones Potential immuno-stimulators in vaccine design. Hum. Vaccines Immunother. 2013, 9, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sot, B.; Rubio-Munoz, A.; Leal-Quintero, A.; Martinez-Sabando, J.; Marcilla, M.; Roodveldt, C.; Valpuesta, J.M. The chaperonin CCT inhibits assembly of alpha-synuclein amyloid fibrils by a specific, conformation-dependent interaction. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.M.; Joachimiak, L.A.; Tan, Z.Q.; Tomlinson, A.; Housman, D.E.; Glabe, C.G.; Potkin, S.G.; Frydman, J.; Thompson, L.M. Exogenous delivery of chaperonin subunit fragment ApiCCT1 modulates mutant Huntingtin cellular phenotypes. Proc. Natl. Acad. Sci. USA 2013, 110, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Ishida, R.; Okamoto, T.; Motojima, F.; Kubota, H.; Takahashi, H.; Tanabe, M.; Oka, T.; Kitamura, A.; Kinjo, M.; Yoshida, M.; et al. Physicochemical Properties of the Mammalian Molecular Chaperone HSP60. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Okamoto, T.; Yamamoto, H.; Kudo, I.; Matsumoto, K.; Odaka, M.; Grave, E.; Itoh, H. HSP60 possesses a GTPase activity and mediates protein folding with HSP10. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.H.; Li, G.J.; Davis, J.; Zhu, D.; Wang, Y.; Pan, C.; Zhang, J. Identification of novel proteins associated with both alpha-synuclein and DJ-1. Mol. Cell. Proteom. 2007, 6, 845–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakovic, A.; Grunewald, A.; Voges, L.; Hofmann, S.; Orolicki, S.; Lohmann, K.; Klein, C. PINK1-Interacting Proteins: Proteomic Analysis of Overexpressed PINK1. Parkinsons Dis. 2011. [Google Scholar] [CrossRef] [Green Version]
- Davison, E.J.; Pennington, K.; Hung, C.C.; Peng, J.H.; Rafiq, R.; Ostareck-Lederer, A.; Ostareck, D.H.; Ardley, H.C.; Banks, R.E.; Robinson, P.A. Proteomic analysis of increased Parkin expression and its interactants provides evidence for a role in modulation of mitochondrial function. Proteomics 2009, 9, 4284–4297. [Google Scholar] [CrossRef]
- Zhao, C.; Li, H.; Zhao, X.J.; Liu, Z.X.; Zhou, P.; Liu, Y.; Feng, M.J. Heat shock protein 60 affects behavioral improvement in a rat model of Parkinson’s disease grafted with human umbilical cord mesenchymal stem cell-derived dopaminergic-like neurons. Neurochem. Res. 2016, 41, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Monti, C.; Bondi, H.; Urbani, A.; Fasano, M.; Alberio, T. Systems biology analysis of the proteomic alterations induced by MPP+, a Parkinson’s disease-related mitochondrial toxin. Front. Cell. Neurosci. 2015, 9, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangione, M.R.; Vilasi, S.; Marino, C.; Librizzi, F.; Canale, C.; Spigolon, D.; Bucchieri, F.; Fucarino, A.; Passantino, R.; Cappello, F.; et al. Hsp60, amateur chaperone in amyloid-beta fibrillogenesis. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Fukui, N.; Araki, K.; Hongo, K.; Mizobata, T.; Kawata, Y. Modulating the Effects of the Bacterial Chaperonin GroEL on Fibrillogenic Polypeptides through Modification of Domain Hinge Architecture. J. Biol. Chem. 2016, 291, 25217–25226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojha, B.; Fukui, N.; Hongo, K.; Mizobata, T.; Kawata, Y. Suppression of amyloid fibrils using the GroEL apical domain. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Lo Bianco, C.; Shorter, J.; Regulier, E.; Lashuel, H.; Iwatsubo, T.; Lindquist, S.; Aebischer, P. Hsp104 antagonizes alpha-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J. Clin. Investig. 2008, 118, 3087–3097. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.C.; Carroni, M.; Nussbaum-Krammer, C.; Mogk, A.; Nillegoda, N.B.; Szlachcic, A.; Guilbride, D.L.; Saibil, H.R.; Mayer, M.P.; Bukau, B. Human Hsp70 Disaggregase Reverses Parkinson’s-Linked alpha-Synuclein Amyloid Fibrils. Mol. Cell 2015, 59, 781–793. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.; Whiten, D.R.; Brown, J.W.P.; Horrocks, M.H.; San Gil, R.; Dobson, C.M.; Klenerman, D.; van Oijen, A.M.; Ecroyd, H. The small heat shock protein Hsp27 binds alpha-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 2018, 293, 4486–4497. [Google Scholar] [CrossRef] [Green Version]
- Chaari, A.; Hoarau-Vechot, J.; Ladjimi, M. Applying chaperones to protein-misfolding disorders: Molecular chaperones against alpha-synuclein in Parkinson’s disease (vol 60, pg 196, 2013). Int. J. Biol. Macromol. 2014, 68, 274. [Google Scholar] [CrossRef]
- Machida, K.; Fujiwara, R.; Tanaka, T.; Sakane, I.; Hongo, K.; Mizobata, T.; Kawata, Y. Gly192 at hinge 2 site in the chaperonin GroEL plays a pivotal role in the dynamic apical domain movement that leads to GroES binding and efficient encapsulation of substrate proteins. Biochim. Biophys. Acta Proteins Proteom. 2009, 1794, 1344–1354. [Google Scholar] [CrossRef]
- Parnas, A.; Nisemblat, S.; Weiss, C.; Levy-Rimler, G.; Pri-Or, A.; Zor, T.; Lund, P.A.; Bross, P.; Azem, A. Identification of Elements That Dictate the Specificity of Mitochondrial Hsp60 for Its Co-Chaperonin. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Someya, Y.; Yui, H. Fluorescence Lifetime Probe for Solvent Microviscosity Utilizing Anilinonaphthalene Sulfonate. Anal. Chem. 2010, 82, 5470–5476. [Google Scholar] [CrossRef] [PubMed]
- Bharat, S.V.; Shekhtman, A.; Pande, J. The cataract-associated V41M mutant of human gamma S-crystallin shows specific structural changes that directly enhance local surface hydrophobicity. Biochem. Biophys. Res. Commun. 2014, 443, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Enriquez, A.S.; Rojo, H.M.; Bhatt, J.M.; Molugu, S.K.; Hildenbrand, Z.L.; Bernal, R.A. The human mitochondrial Hsp60 in the APO conformation forms a stable tetradecameric complex. Cell Cycle 2017, 16, 1309–1319. [Google Scholar] [CrossRef]
- Gong, H.; He, Z.H.; Peng, A.L.; Zhang, X.; Cheng, B.A.; Sun, Y.; Zheng, L.; Huang, K. Effects of several quinones on insulin aggregation. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Fenton, W.A.; Kashi, Y.; Furtak, K.; Horwich, A.L. Residues in chaperonin GroEL required for polypeptide binding and release. Nature 1994, 371, 614–619. [Google Scholar] [CrossRef]
- Chen, L.L.; Sigler, P.B. The crystal structure of a GroEL/peptide complex: Plasticity as a basis for substrate diversity. Cell 1999, 99, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Noi, K.; Kitamura, A.; Hirai, H.; Hongo, K.; Sakurai, T.; Mizobata, T.; Kawata, Y. Suppression of Sup35 amyloid fibril formation by group II chaperonin from Thermoplasma acidophilum. Am. J. Mol. Biol. 2012, 2, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Altamirano, M.M.; Golbik, R.; Zahn, R.; Buckle, A.M.; Fersht, A.R. Refolding chromatography with immobilized mini-chaperones. Proc. Natl. Acad. Sci. USA 1997, 94, 3576–3578. [Google Scholar] [CrossRef] [Green Version]
- Tam, S.; Geller, R.; Spiess, C.; Frydman, J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 2006, 8, 1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantoliano, M.W.; Ladner, R.C.; Bryan, P.N.; Rollence, M.L.; Wood, J.F.; Poulos, T.L. Protein engineering of subtilisin BPN’—enhanced stabilization through the introduction of 2 cysteines to form a disulfide bond. Biochemistry 1987, 26, 2077–2082. [Google Scholar] [CrossRef] [PubMed]
- Jenke, D.R.; Brown, D.S. Determination of cysteine in pharmaceuticals via liquid-chromatography with postcolumn derivatization. Anal. Chem. 1987, 59, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Wien, F.; Bulyaki, E.; Kun, J.; Moussong, E.; Lee, Y.H.; Goto, Y.; Refregiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef] [PubMed]
- Abriata, L.A. A Simple Spreadsheet Program To Simulate and Analyze the Far-UV Circular Dichroism Spectra of Proteins. J. Chem. Educ. 2011, 88, 1268–1273. [Google Scholar] [CrossRef]
- Ungerstedt, U. 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur. J. Pharmacol. 1968, 5, 107–110. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Sonderby, C.K.; Sonderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef]
- Kim, S.A.; Chang, S.; Yoon, J.H.; Ahn, S.G. TAT-Hsp40 inhibits oxidative stress-mediated cytotoxicity via the inhibition of Hsp70 ubiquitination. FEBS Lett. 2008, 582, 734–740. [Google Scholar] [CrossRef] [Green Version]
- Nagel, F.; Dohm, C.P.; Bahr, M.; Wouters, F.S.; Dietz, G.P.H. Quantitative evaluation of chaperone activity and neuroprotection by different preparations of a cell-penetrating Hsp70. J. Neurosci. Methods 2008, 171, 226–232. [Google Scholar] [CrossRef]
- Nagel, F.; Falkenburger, B.H.; Tonges, L.; Kowsky, S.; Poppelmeyer, C.; Schulz, J.B.; Bahr, M.; Dietz, G.P.H. Tat-Hsp70 protects dopaminergic neurons in midbrain cultures and in the substantia nigra in models of Parkinson’s disease. J. Neurochem. 2008, 105, 853–864. [Google Scholar] [CrossRef]
- Doeppner, T.R.; Ewert, T.A.S.; Tonges, L.; Herz, J.; Zechariah, A.; ElAli, A.; Ludwig, A.K.; Giebel, B.; Nagel, F.; Dietz, G.P.H.; et al. Transduction of Neural Precursor Cells with TAT-Heat Shock Protein 70 Chaperone: Therapeutic Potential Against Ischemic Stroke after Intrastriatal and Systemic Transplantation. Stem Cells 2012, 30, 1297–1310. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Yagi, H.; Kusaka, E.; Hongo, K.; Mizobata, T.; Kawata, Y. Amyloid fibril formation of alpha-synuclein is accelerated by preformed amyloid seeds of other proteins—Implications for the mechanism of transmissible conformational diseases. J. Biol. Chem. 2005, 280, 38609–38616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, T.; Cepko, C.L. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2004, 101, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Kd (nM) | kon (M−1s−1) | koff (s−1) |
|---|---|---|---|
| Hsp60 WT | 4.29 | 2.30 × 105 | 9.89 × 10−4 |
| Hsp60 GW | 0.963 | 3.64 × 105 | 3.51 × 10−4 |
| Sample | Kd (nM) | kon (M−1s−1) | koff (s−1) |
|---|---|---|---|
| Hsp60 AD(Cys) | 5.04 | 5.85 × 105 | 2.95 × 10−3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, H.; Fukui, N.; Adachi, M.; Saiki, E.; Yamasaki, A.; Matsumura, R.; Kuroyanagi, D.; Hongo, K.; Mizobata, T.; Kawata, Y. Human Molecular Chaperone Hsp60 and Its Apical Domain Suppress Amyloid Fibril Formation of α-Synuclein. Int. J. Mol. Sci. 2020, 21, 47. https://doi.org/10.3390/ijms21010047
Yamamoto H, Fukui N, Adachi M, Saiki E, Yamasaki A, Matsumura R, Kuroyanagi D, Hongo K, Mizobata T, Kawata Y. Human Molecular Chaperone Hsp60 and Its Apical Domain Suppress Amyloid Fibril Formation of α-Synuclein. International Journal of Molecular Sciences. 2020; 21(1):47. https://doi.org/10.3390/ijms21010047
Chicago/Turabian StyleYamamoto, Hanae, Naoya Fukui, Mayuka Adachi, Eiichi Saiki, Anna Yamasaki, Rio Matsumura, Daichi Kuroyanagi, Kunihiro Hongo, Tomohiro Mizobata, and Yasushi Kawata. 2020. "Human Molecular Chaperone Hsp60 and Its Apical Domain Suppress Amyloid Fibril Formation of α-Synuclein" International Journal of Molecular Sciences 21, no. 1: 47. https://doi.org/10.3390/ijms21010047
APA StyleYamamoto, H., Fukui, N., Adachi, M., Saiki, E., Yamasaki, A., Matsumura, R., Kuroyanagi, D., Hongo, K., Mizobata, T., & Kawata, Y. (2020). Human Molecular Chaperone Hsp60 and Its Apical Domain Suppress Amyloid Fibril Formation of α-Synuclein. International Journal of Molecular Sciences, 21(1), 47. https://doi.org/10.3390/ijms21010047