Detection of Loss of Heterozygosity in cfDNA of Advanced EGFR- or KRAS-Mutated Non-Small-Cell Lung Cancer Patients

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
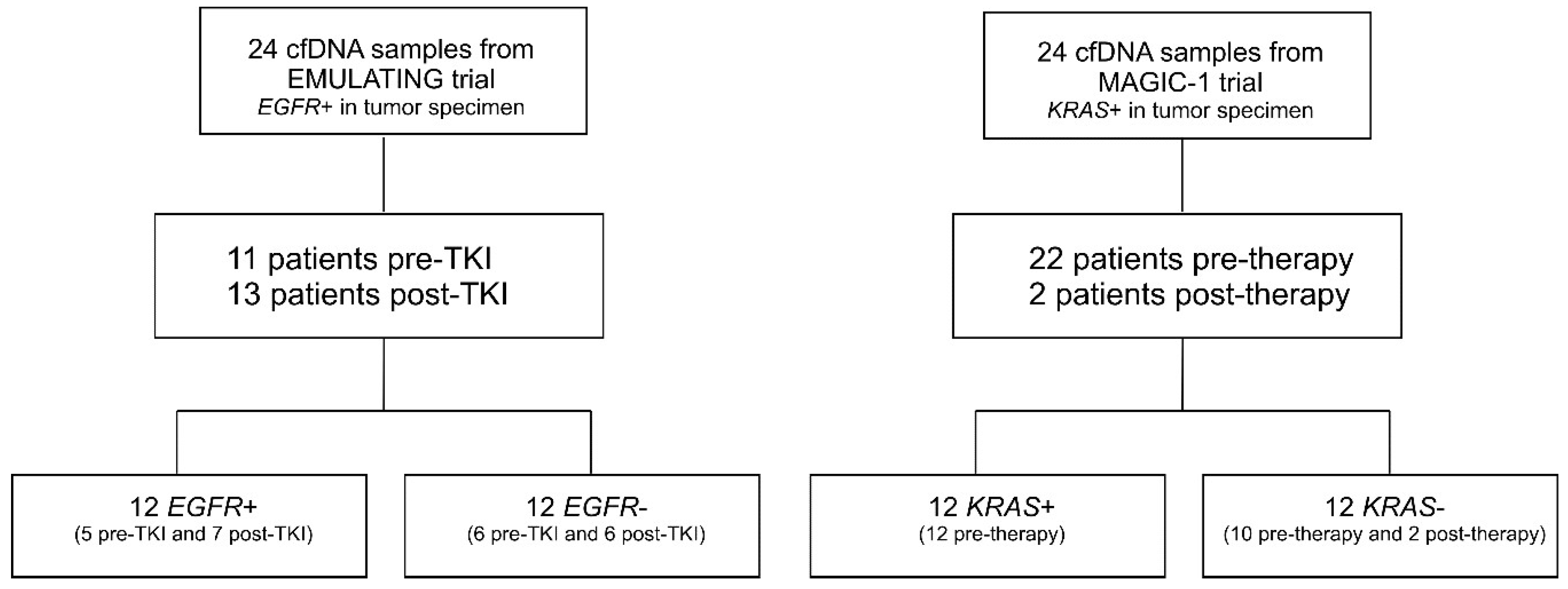
2.1. Patients
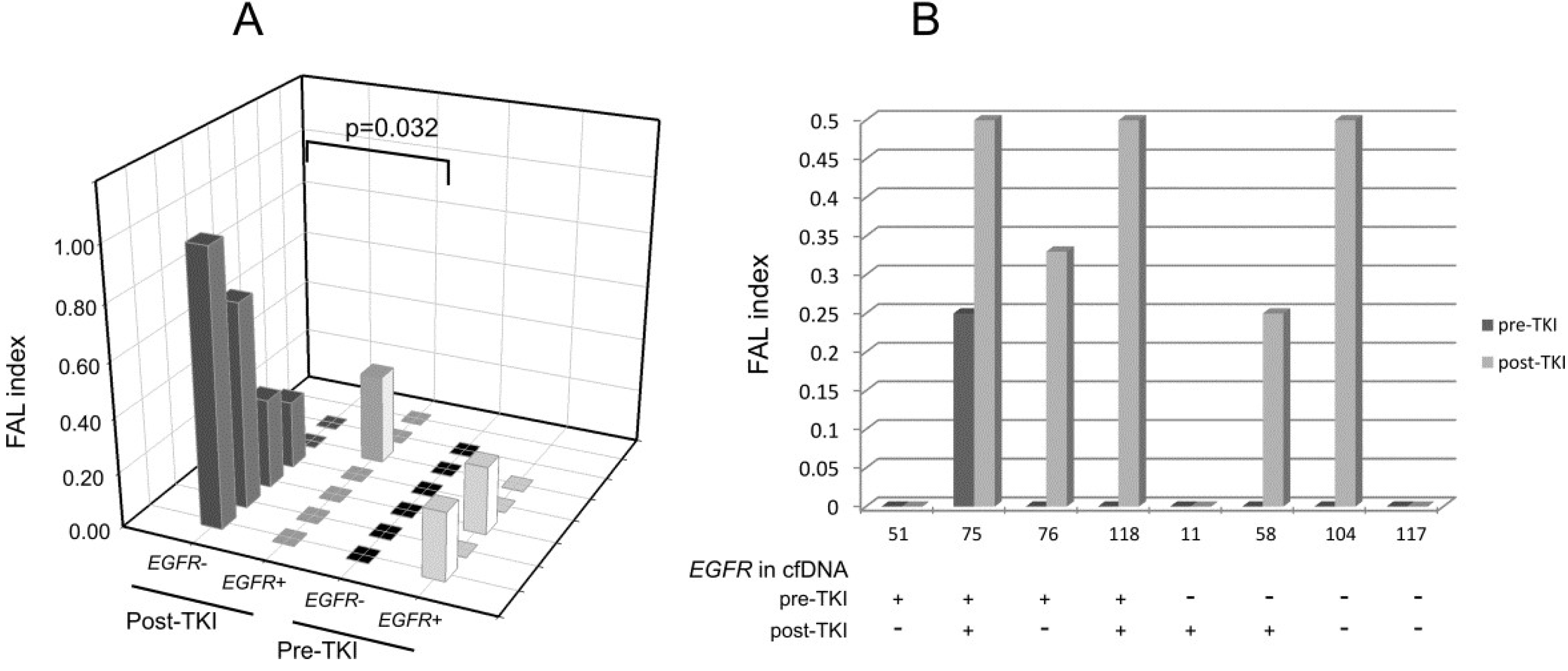
2.2. Analysis of LOH in EGFR-Mutated Patients
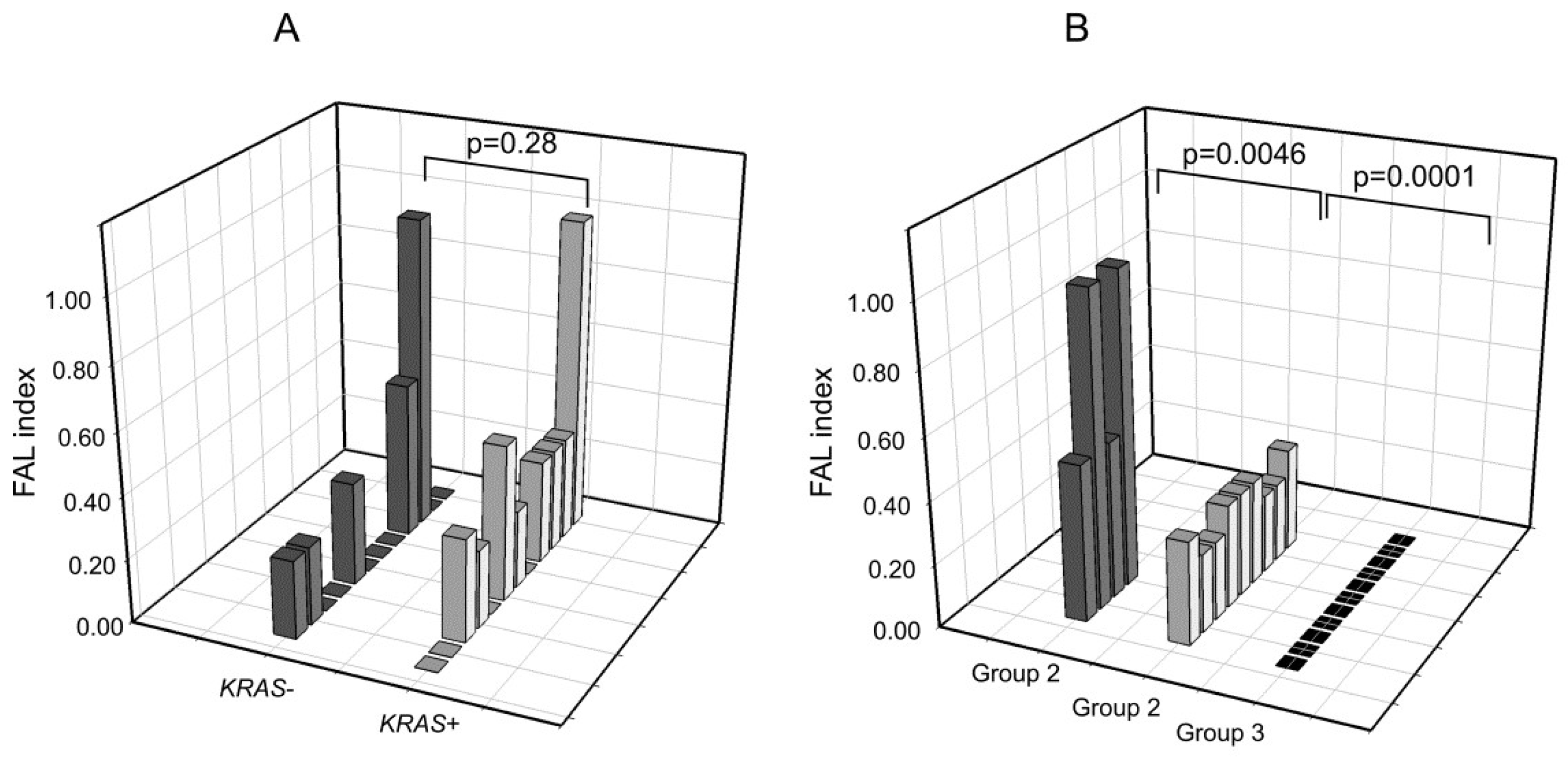
2.3. Analysis of LOH in KRAS-Mutated Patients
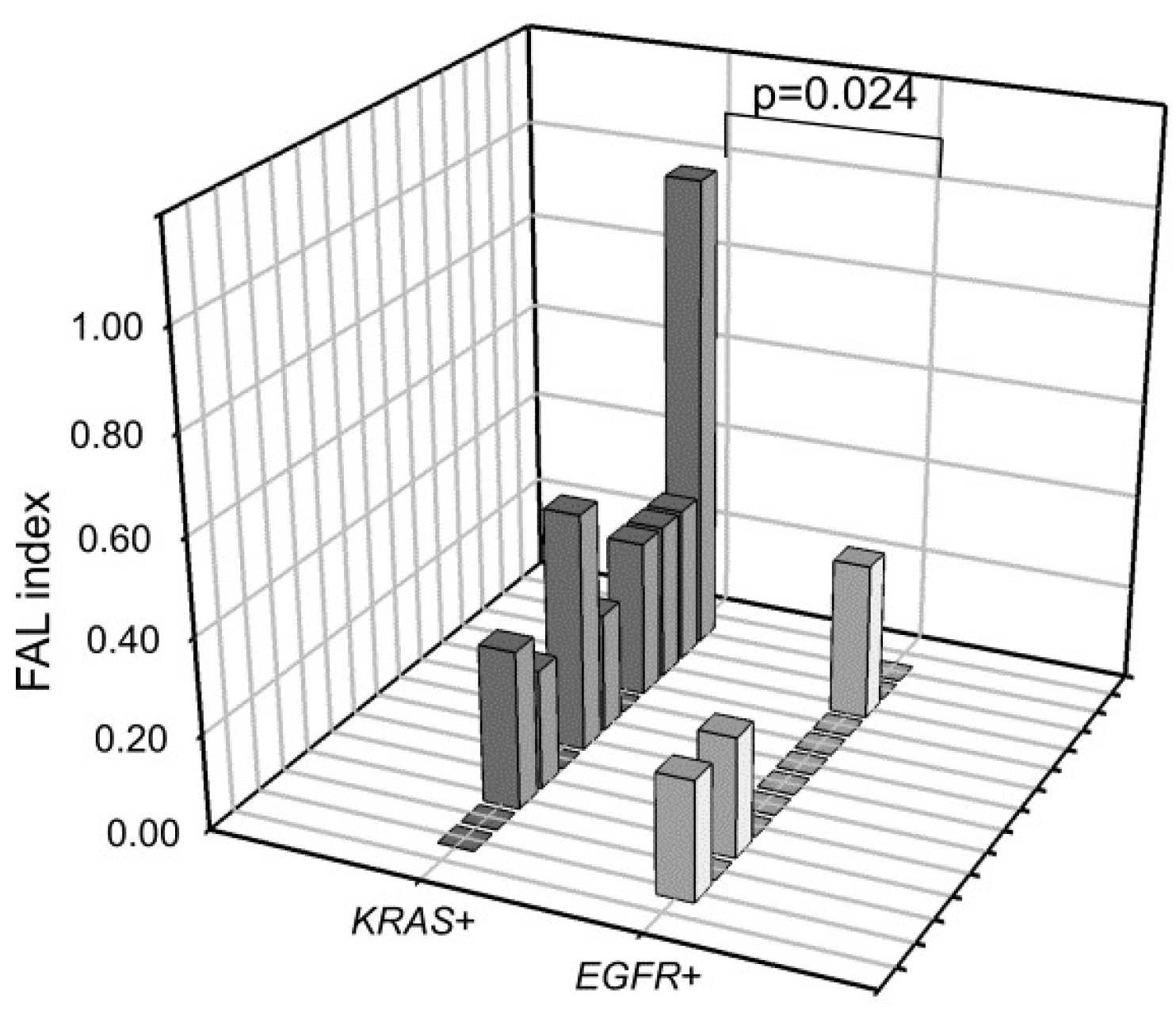
2.4. LOH Frequency Comparison between EGFR+ and KRAS+ cfDNA Samples
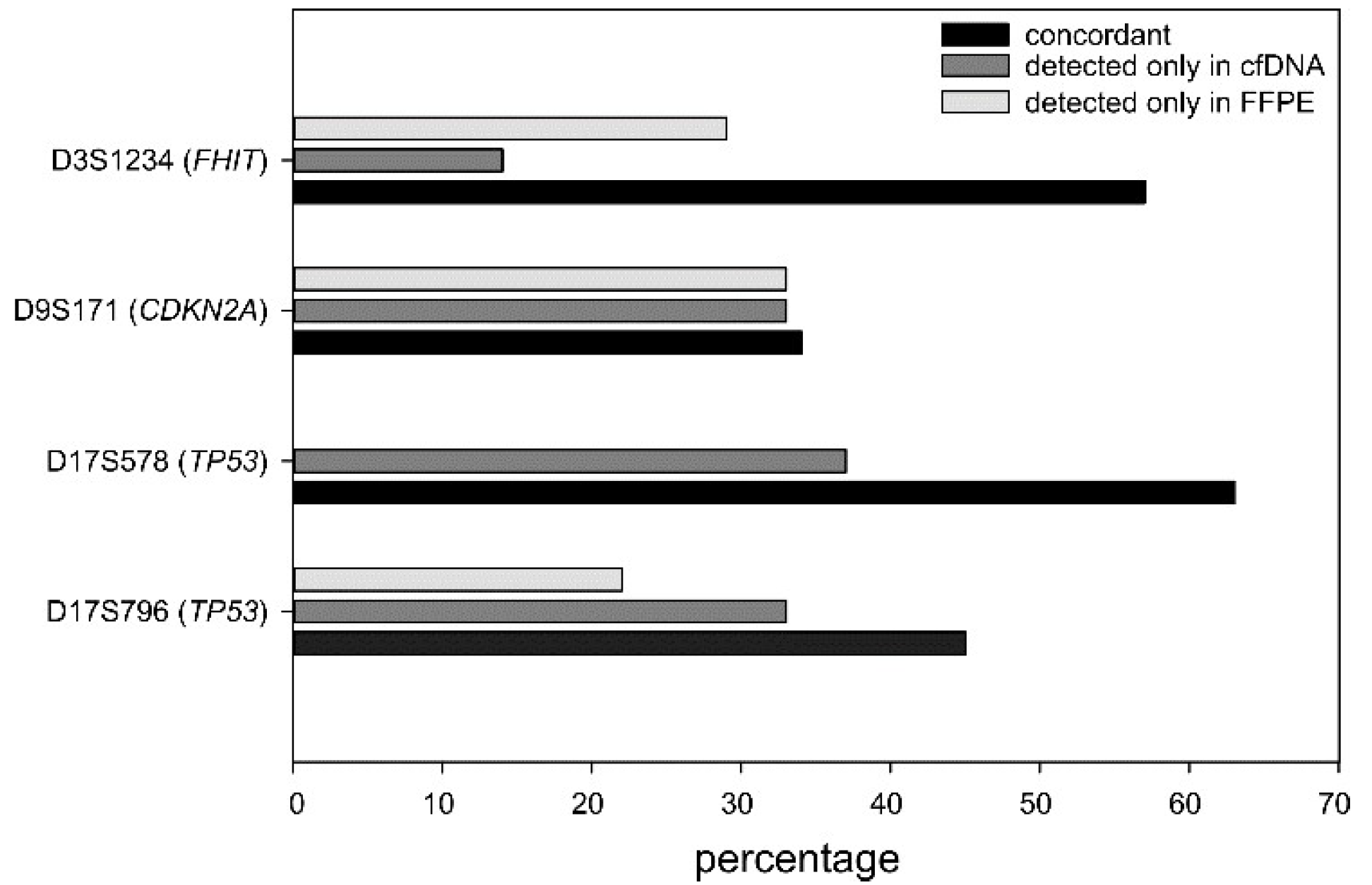
2.5. LOH Comparison between Plasma and Tumor Specimens
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Sample Collection and DNA Extraction
4.3. DNA Extraction
4.4. Genetic Analyses
4.5. LOH and MSI Analysis
4.6. Data Analysis and Statistics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cheng, T.Y.; Cramb, S.M.; Baade, P.D.; Youlden, D.R.; Nwogu, C.; Reid, M.E. The International Epidemiology of Lung Cancer: Latest Trends, Disparities, and Tumor Characteristics. J. Thorac. Oncol. 2016, 11, 1653–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meador, C.B.; Micheel, C.M.; Levy, M.A.; Lovly, C.M.; Horn, L.; Warner, J.L.; Johnson, D.B.; Zhao, Z.; Anderson, I.A.; Sosman, J.A.; et al. Beyond histology: Translating tumor genotypes into clinically effective targeted therapies. Clin. Cancer Res. 2014, 20, 2264–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, W.; Girard, N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12, 175–180. [Google Scholar] [CrossRef]
- Barlesi, F.; Mazieres, J.; Merlio, J.P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.; Besse, B.; Rouquette, I.; Westeel, V.; et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016, 387, 1415–1426. [Google Scholar] [CrossRef]
- Kempf, E.; Rousseau, B.; Besse, B.; Paz-Ares, L. KRAS oncogene in lung cancer: Focus on molecularly driven clinical trials. Eur. Respir. Rev. 2016, 25, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Hagemann, I.S.; Devarakonda, S.; Lockwood, C.M.; Spencer, D.H.; Guebert, K.; Bredemeyer, A.J.; Al-Kateb, H.; Nguyen, T.T.; Duncavage, E.J.; Cottrell, C.E.; et al. Clinical next-generation sequencing in patients with non-small cell lung cancer. Cancer 2015, 121, 631–639. [Google Scholar] [CrossRef]
- Rolfo, C.; Mack, P.C.; Scagliotti, G.V.; Baas, P.; Barlesi, F.; Bivona, T.G.; Herbst, R.S.; Mok, T.S.; Peled, N.; Pirker, R.; et al. Liquid Biopsy for Advanced Non-Small Cell Lung Cancer (NSCLC): A Statement Paper from the IASLC. J. Thorac. Oncol. 2018, 13, 1248–1268. [Google Scholar] [CrossRef] [Green Version]
- Mayo-de-Las-Casas, C.; Garzon Ibanez, M.; Jordana-Ariza, N.; Garcia-Pelaez, B.; Balada-Bel, A.; Villatoro, S.; Malapelle, U.; Karachaliou, N.; Troncone, G.; Rosell, R.; et al. An update on liquid biopsy analysis for diagnostic and monitoring applications in non-small cell lung cancer. Expert. Rev. Mol. Diagn. 2018, 18, 35–45. [Google Scholar] [CrossRef]
- Normanno, N.; Denis, M.G.; Thress, K.S.; Ratcliffe, M.; Reck, M. Guide to detecting epidermal growth factor receptor (EGFR) mutations in ctDNA of patients with advanced non-small-cell lung cancer. Oncotarget 2017, 8, 12501–12516. [Google Scholar] [CrossRef] [Green Version]
- Reck, M.; Hagiwara, K.; Han, B.; Tjulandin, S.; Grohe, C.; Yokoi, T.; Morabito, A.; Novello, S.; Arriola, E.; Molinier, O.; et al. ctDNA Determination of EGFR Mutation Status in European and Japanese Patients with Advanced NSCLC: The ASSESS Study. J. Thorac. Oncol. 2016, 11, 1682–1689. [Google Scholar] [CrossRef] [Green Version]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldrin, E.; Rumiato, E.; Fassan, M.; Cappellesso, R.; Rugge, M.; Chiarion-Sileni, V.; Ruol, A.; Alfieri, R.; Cagol, M.; Castoro, C.; et al. Genetic features of metachronous esophageal cancer developed in Hodgkin’s lymphoma or breast cancer long-term survivors: An exploratory study. PLoS ONE 2015, 10, e0117070. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Shaw, J.A.; Walker, R.A. Sporadic breast cancer in young women: Prevalence of loss of heterozygosity at p53, BRCA1 and BRCA2. Int. J. Cancer 2002, 98, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Thiagalingam, S.; Laken, S.; Willson, J.K.; Markowitz, S.D.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Mechanisms underlying losses of heterozygosity in human colorectal cancers. Proc. Natl. Acad. Sci. USA 2001, 98, 2698–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.P.; Wu, D.Y.; Xu, L.; Xin, Y. Loss of heterozygosity and microsatellite instabilities of fragile histidine triad gene in gastric carcinoma. World J. Gastroenterol. 2006, 12, 3766–3769. [Google Scholar] [CrossRef]
- Beau-Faller, M.; Gaub, M.P.; Schneider, A.; Ducrocq, X.; Massard, G.; Gasser, B.; Chenard, M.P.; Kessler, R.; Anker, P.; Stroun, M.; et al. Plasma DNA microsatellite panel as sensitive and tumor-specific marker in lung cancer patients. Int. J. Cancer 2003, 105, 361–370. [Google Scholar] [CrossRef]
- Virmani, A.K.; Fong, K.M.; Kodagoda, D.; McIntire, D.; Hung, J.; Tonk, V.; Minna, J.D.; Gazdar, A.F. Allelotyping demonstrates common and distinct patterns of chromosomal loss in human lung cancer types. Genes Chromosomes Cancer 1998, 21, 308–319. [Google Scholar] [CrossRef]
- Ryland, G.L.; Doyle, M.A.; Goode, D.; Boyle, S.E.; Choong, D.Y.; Rowley, S.M.; Li, J.; Bowtell, D.D.; Tothill, R.W.; Campbell, I.G.; et al. Loss of heterozygosity: What is it good for? BMC Med. Genom. 2015, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Mansuet-Lupo, A.; Barritault, M.; Alifano, M.; Janet-Vendroux, A.; Zarmaev, M.; Biton, J.; Velut, Y.; Le Hay, C.; Cremer, I.; Regnard, J.F.; et al. Proposal for a Combined Histomolecular Algorithm to Distinguish Multiple Primary Adenocarcinomas from Intrapulmonary Metastasis in Patients with Multiple Lung Tumors. J. Thorac. Oncol. 2019, 14, 844–856. [Google Scholar] [CrossRef]
- Murphy, S.J.; Harris, F.R.; Kosari, F.; Barreto Siqueira Parrilha Terra, S.; Nasir, A.; Johnson, S.H.; Serla, V.; Smadbeck, J.B.; Halling, G.C.; Karagouga, G.; et al. Using Genomics to Differentiate Multiple Primaries From Metastatic Lung Cancer. J. Thorac. Oncol. 2019, 14, 1567–1582. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Bao, H.; Le, X.; Fan, X.; Tang, M.; Shi, X.; Zhao, J.; Yan, J.; Xu, Y.; Quek, K.; et al. Distinct co-acquired alterations and genomic evolution during TKI treatment in non-small-cell lung cancer patients with or without acquired T790M mutation. Oncogene 2019. [Google Scholar] [CrossRef]
- Rumiato, E.; Boldrin, E.; Malacrida, S.; Realdon, S.; Fassan, M.; Morbin, T.; Battaglia, G.; Amadori, A.; Rugge, M.; Saggioro, D. Detection of genetic alterations in cfDNA as a possible strategy to monitor the neoplastic progression of Barrett’s esophagus. Transl. Res. 2017, 190, 16–24.e1. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, C.F.; Stoecklein, N.H.; Jazra, S.; Hosch, S.B.; Peiper, M.; Scheunemann, P.; Am Esch, J.S.; Knoefel, W.T. The detection of oesophageal adenocarcinoma by serum microsatellite analysis. Eur. J. Surg. Oncol. 2006, 32, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Larson, P.S.; Schlechter, B.L.; Zahid, N.; Finnemore, E.; de las Morenas, A.; Blanchard, R.A.; Rosenberg, C.L. Loss of heterozygosity in serial plasma DNA samples during follow-up of women with breast cancer. Int. J. Cancer 2003, 106, 923–929. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) Clinical Features of EGFR-Mutated Patients | ||
| Age at Diagnosis (years) | Median (range) | 70 (43–87) |
| Gender | Male | 13 (54%) |
| Female | 11 (46%) | |
| Smoking | No | 13 (54.2%) |
| Current | 0 (0%) | |
| Former | 11 (45.8%) | |
| PS | 0 | 6 (25%) |
| 1 | 18 (75%) | |
| 2 | 0 (0%) | |
| Stage at Diagnosis | I–II | 0 (0%) |
| III–IV | 24 (100%) | |
| cfDNA EGFR test | Diagnosis | 11 (50%) |
| Progression Disease | 13 (50%) | |
| Total | 24 | |
| (B) Clinical Features of KRAS-Mutated Patients | ||
| Age at Diagnosis (years) | Median (range) | 69.5 (48–85) |
| Gender | Male | 11 (46%) |
| Female | 13 (54%) | |
| Smoking | No | 4 (16.7%) |
| Current | 9 (37.5%) | |
| Former | 11 (45.8%) | |
| PS | 0 | 8 (33%) |
| 1 | 15 (63%) | |
| 2 | 1 (4 %) | |
| Stage at Diagnosis | I–II | 0 (0%) |
| III–IV | 24 (100%) | |
| cfDNA KRAS test | Diagnosis | 22 (92%) |
| Progression Disease | 2 (8%) | |
| Total | 24 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boldrin, E.; Nardo, G.; Zulato, E.; Bonanno, L.; Polo, V.; Frega, S.; Pavan, A.; Indraccolo, S.; Saggioro, D. Detection of Loss of Heterozygosity in cfDNA of Advanced EGFR- or KRAS-Mutated Non-Small-Cell Lung Cancer Patients. Int. J. Mol. Sci. 2020, 21, 66. https://doi.org/10.3390/ijms21010066
Boldrin E, Nardo G, Zulato E, Bonanno L, Polo V, Frega S, Pavan A, Indraccolo S, Saggioro D. Detection of Loss of Heterozygosity in cfDNA of Advanced EGFR- or KRAS-Mutated Non-Small-Cell Lung Cancer Patients. International Journal of Molecular Sciences. 2020; 21(1):66. https://doi.org/10.3390/ijms21010066
Chicago/Turabian StyleBoldrin, Elisa, Giorgia Nardo, Elisabetta Zulato, Laura Bonanno, Valentina Polo, Stefano Frega, Alberto Pavan, Stefano Indraccolo, and Daniela Saggioro. 2020. "Detection of Loss of Heterozygosity in cfDNA of Advanced EGFR- or KRAS-Mutated Non-Small-Cell Lung Cancer Patients" International Journal of Molecular Sciences 21, no. 1: 66. https://doi.org/10.3390/ijms21010066
APA StyleBoldrin, E., Nardo, G., Zulato, E., Bonanno, L., Polo, V., Frega, S., Pavan, A., Indraccolo, S., & Saggioro, D. (2020). Detection of Loss of Heterozygosity in cfDNA of Advanced EGFR- or KRAS-Mutated Non-Small-Cell Lung Cancer Patients. International Journal of Molecular Sciences, 21(1), 66. https://doi.org/10.3390/ijms21010066