Current Projection Methods-Induced Biases at Subgroup Detection for Machine-Learning Based Data-Analysis of Biomedical Data



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
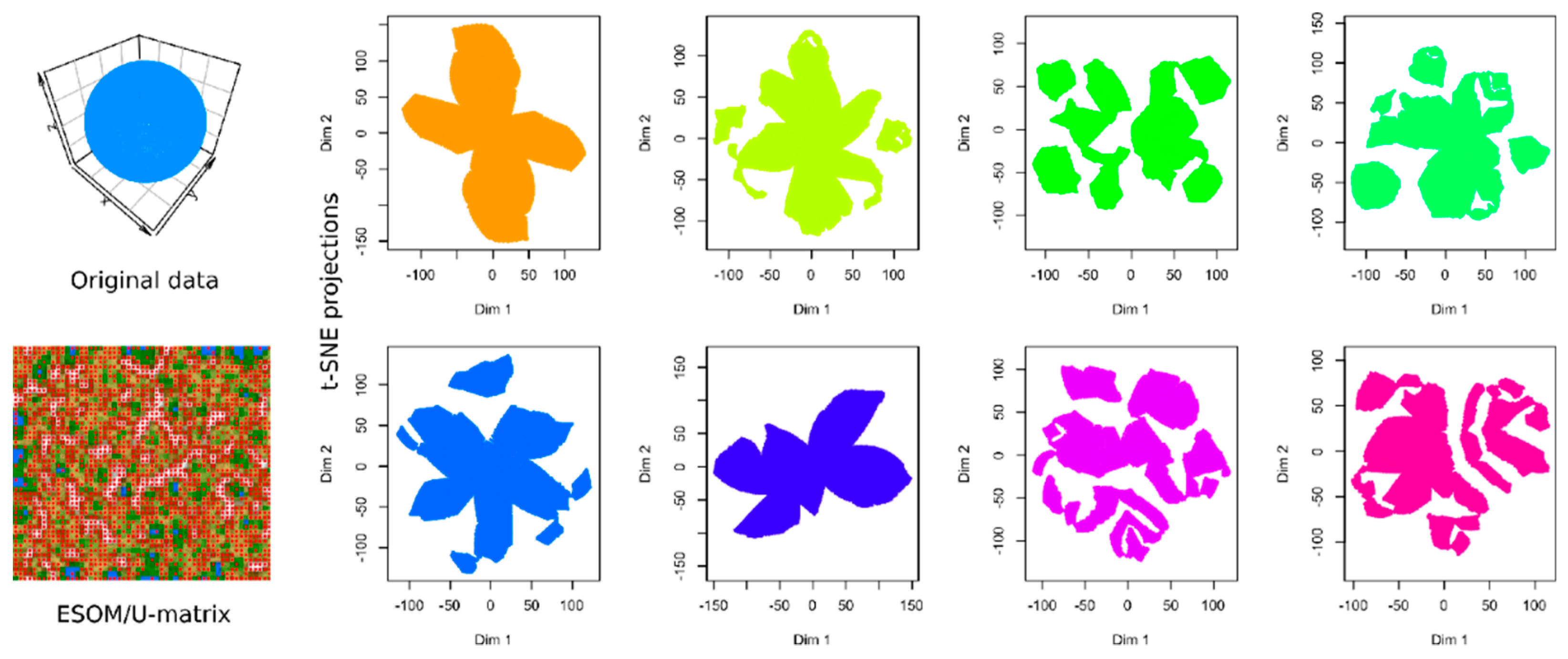
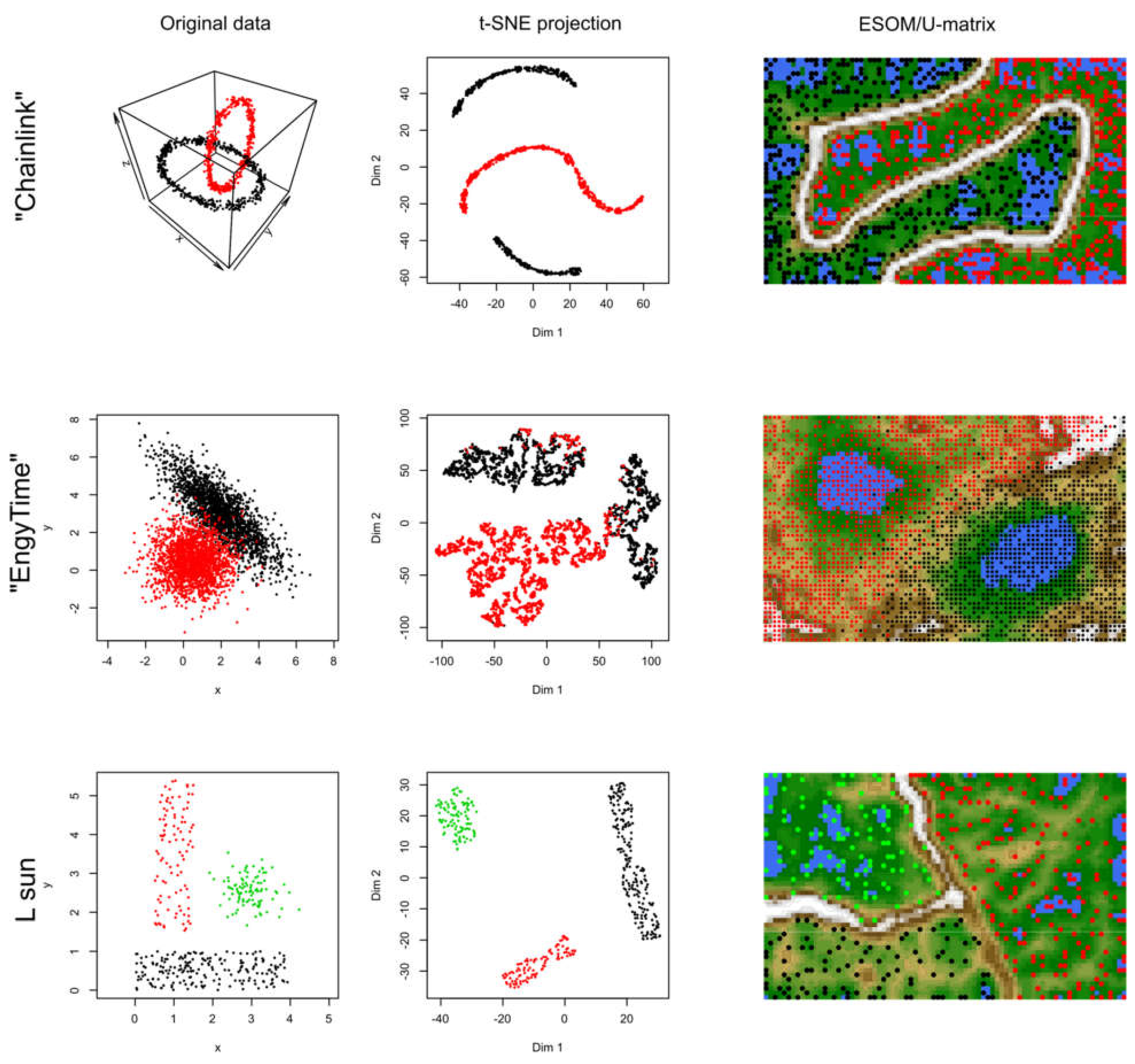
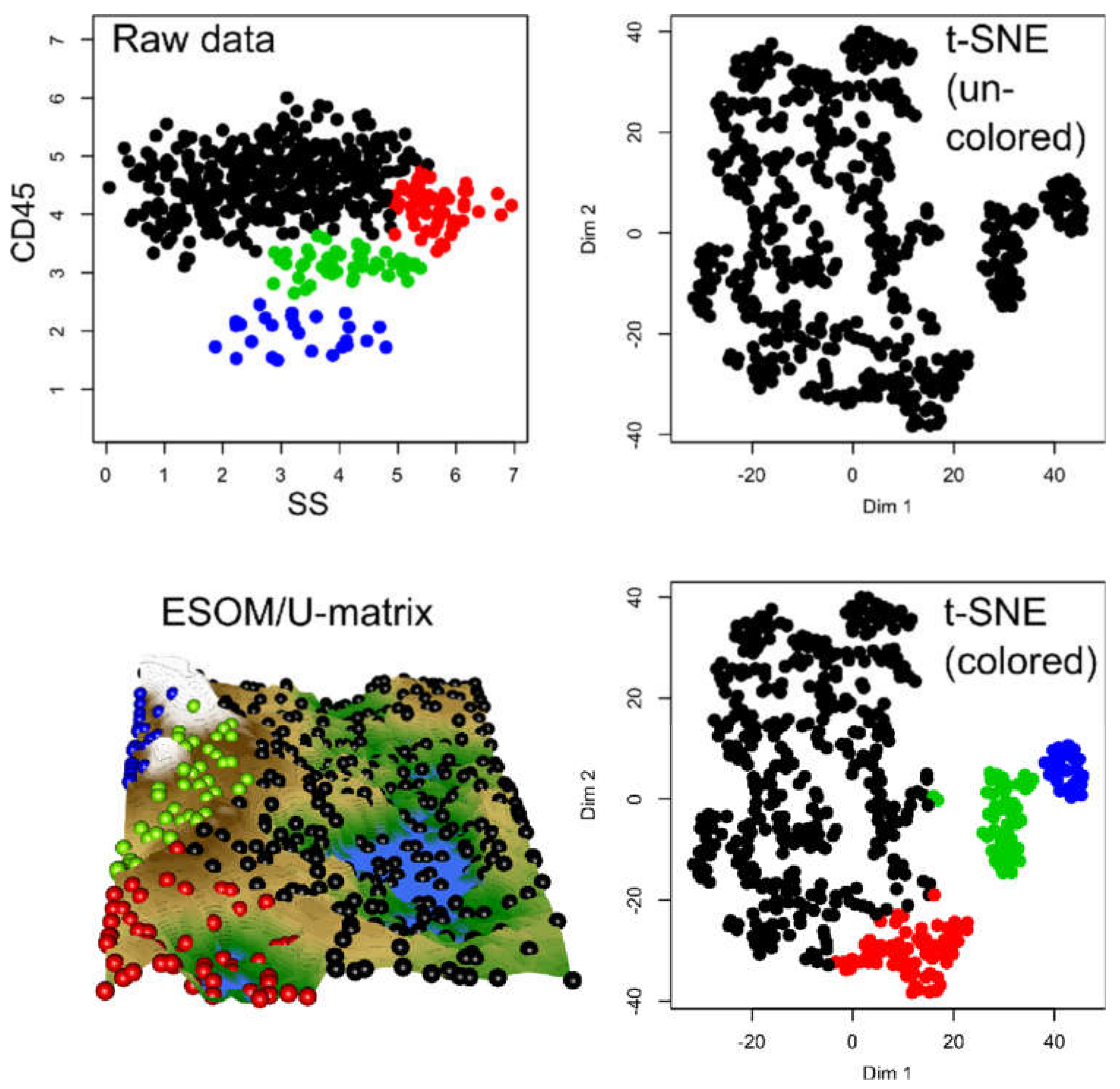
2.1. Results of t-SNE Analysis in Artificial Data Sets
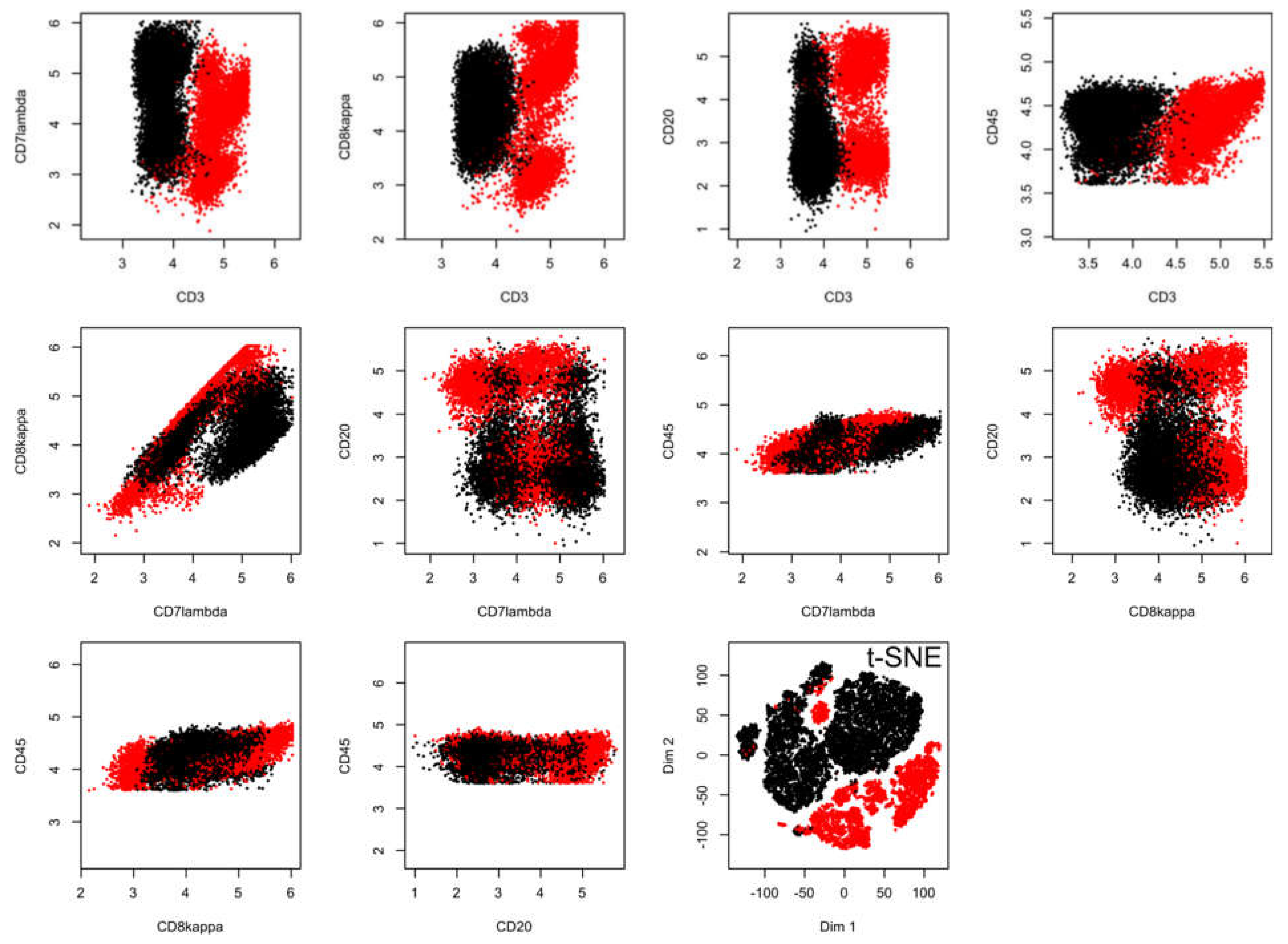
2.2. Results of t-SNE Analysis in Biomedical Data Sets

2.3. Causes of Heterogeneous t-SNE Performance in Different Data Sets
2.4. Effects of Tuning the t-SNE Performance
3. Materials and Methods
3.1. Data Sets
3.2. Data Projection and Subgroup Identification Using t-SNE
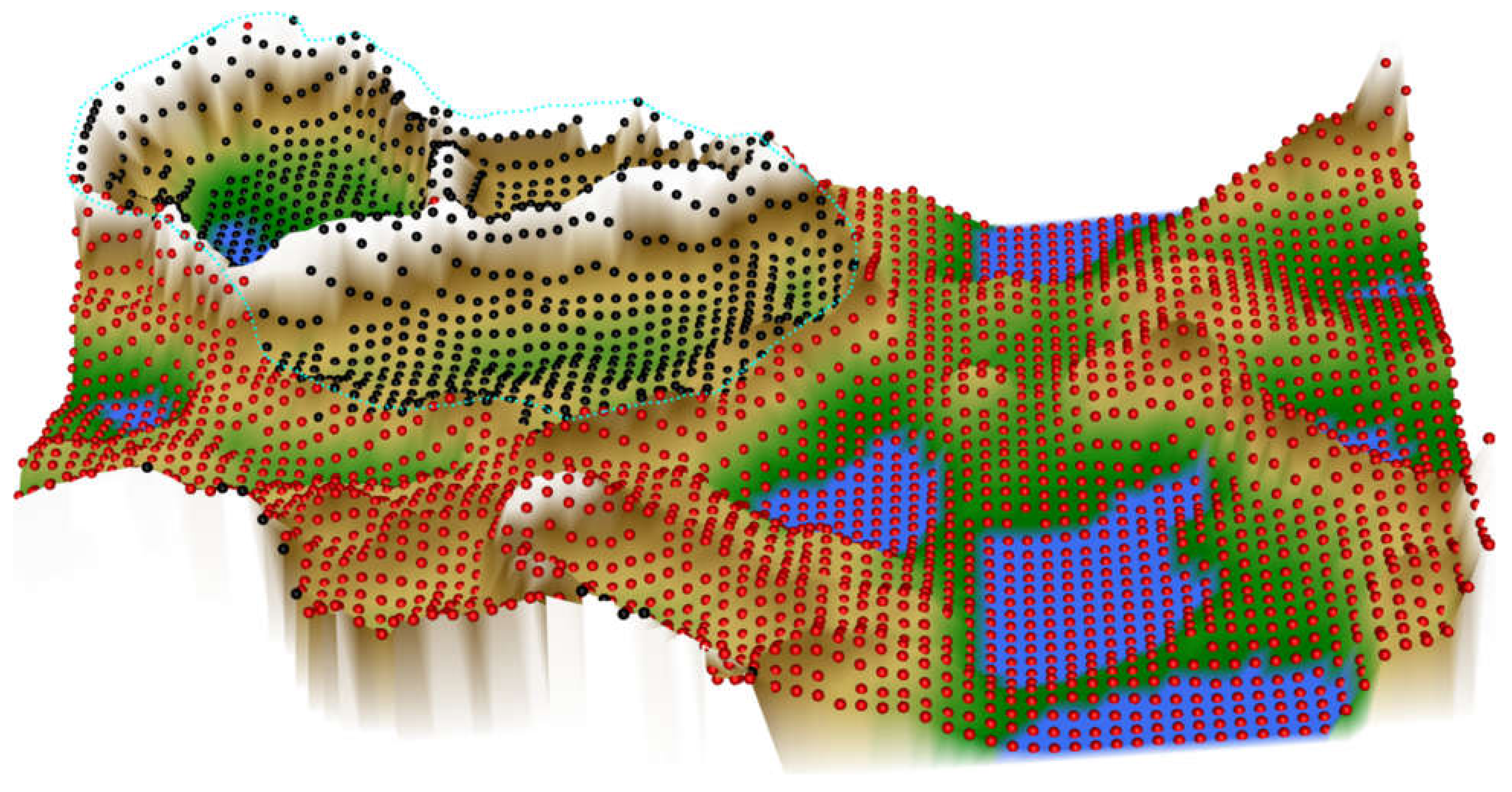
3.3. Data Projection and Subgroup Identification Using ESOM
3.4. Additional Data Projection Techniques
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saeys, Y.; Van Gassen, S.; Lambrecht, B.N. Computational flow cytometry: Helping to make sense of high-dimensional immunology data. Nat. Rev. Immunol. 2016, 16, 449–462. [Google Scholar] [CrossRef]
- Van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn Res. 2008, 9, 2579–2605. [Google Scholar]
- Donaldson, J. tsne: T-Distributed Stochastic Neighbor Embedding for R (t-SNE) (version 0.1-3) R package. 2016. Available online: https://CRAN.R-project.org/package=tsne (accessed on 15 July 2016).
- Lötsch, J.; Lerch, F.; Djaldetti, R.; Tegeder, I.; Ultsch, A. Identification of disease-distinct complex biomarker patterns by means of unsupervised machine-learning using an interactive R toolbox (Umatrix). Big Data Anal. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Ultsch, A.; Lötsch, J. Machine-learned cluster identification in high-dimensional data. J. Biomed. Inform. 2017, 66, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; Grolemund, G. R for Data Science: Import, Tidy, Transform, Visualize, and Model Data; O‘Reilly Media: Sebastopol, CA, USA, 2017. [Google Scholar]
- Ultsch, A. Maps for Visualization of High-Dimensional Data Spaces. In Proceedings of the Workshop on Self-Organizing Maps (WSOM 2003), Kyushu, Japan, 13–16 November 2003; pp. 225–230. [Google Scholar]
- Le, S.; Josse, J.; Husson, F.C. FactoMineR: A Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Lammers, B. ANN2: Artificial Neural Networks for Anomaly Detection. 2019. Available online: https://github.com/bflammers/ANN2 (accessed on 1 May 2019).
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venna, J.; Kaski, S. Local multidimensional scaling. Neural. Netw. 2006, 19, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Statist. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Ultsch, A.; Thrun, M. Credible Visualizations for Planar Projections. In Proceedings of the 12th International Workshop on Self-Organizing Maps and Learning Vector Quantization, Clustering and Data Visualization (WSOM), Nancy, France, 28–30 June 2017; pp. 256–260. [Google Scholar]
- Ultsch, A. Clustering with SOM: U*C. In Proceedings of the Workshop on Self-Organizing Maps, Paris, France, 1 January 2005; pp. 75–82. [Google Scholar]
- Scott, G.D.; Atwater, S.K.; Gratzinger, D.A. Normative data for flow cytometry immunophenotyping of benign lymph nodes sampled by surgical biopsy. J. Clin. Pathol. 2018, 71, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Lö, J.; Ultsch, A. Generative artificial intelligence based algorithm to increase the predictivity of preclinical studies while keeping sample sizes small. In Statistical Computing 2019; Kestler, H.A., Schmid, M., Lausser, L., Fürstberger, A., Eds.; Ulmer Informatik-Bericht: Günzburg, Germany, 2019; pp. 29–30. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 17 August 2018).
- Kohonen, T. Self-organized formation of topologically correct feature maps. Biol. Cybernet. 1982, 43, 59–69. [Google Scholar] [CrossRef]
- Lötsch, J.; Ultsch, A. A machine-learned knowledge discovery method for associating complex phenotypes with complex genotypes. Application to pain. J. Biomed. Inform. 2013, 46, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Van Gassen, S.; Callebaut, B.; Van Helden, M.J.; Lambrecht, B.N.; Demeester, P.; Dhaene, T.; Saeys, Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. A. 2015, 87, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Ultsch, A. Emergence in Self-Organizing Feature Maps. In Proceedings of the 6th International Workshop on Self-Organizing Maps (WSOM ’07), Bielefeld, Germany, 3–6 September 2007; Ritter, H., Haschke, R., Eds.; Neuroinformatics Group: Bielefeld, Germany, 2007. Available online: https://biecoll.ub.uni-bielefeld.de (accessed on 15 July 2016).
- Ultsch, A.; Weingart, M.; Lötsch, J. 3-D printing as a tool for knowledge discovery in high dimensional data spaces. In Statistical Computing; Fürstberger, A., Lausser, L., Kraus, J.M., Schmid, M., Kestler, H.A., Eds.; Universität Ulm, Fakultät für Ingenieurwissenschaften und Informatik, Schloss Reisensburg: Günzburg, Germany, 2015. [Google Scholar]
- Pearson, K. LIII. On lines and planes of closest fit to systems of points in space. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1901, 2, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning representations by back-propagating errors. Nature 1986, 323, 533–536. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lötsch, J.; Ultsch, A. Current Projection Methods-Induced Biases at Subgroup Detection for Machine-Learning Based Data-Analysis of Biomedical Data. Int. J. Mol. Sci. 2020, 21, 79. https://doi.org/10.3390/ijms21010079
Lötsch J, Ultsch A. Current Projection Methods-Induced Biases at Subgroup Detection for Machine-Learning Based Data-Analysis of Biomedical Data. International Journal of Molecular Sciences. 2020; 21(1):79. https://doi.org/10.3390/ijms21010079
Chicago/Turabian StyleLötsch, Jörn, and Alfred Ultsch. 2020. "Current Projection Methods-Induced Biases at Subgroup Detection for Machine-Learning Based Data-Analysis of Biomedical Data" International Journal of Molecular Sciences 21, no. 1: 79. https://doi.org/10.3390/ijms21010079
APA StyleLötsch, J., & Ultsch, A. (2020). Current Projection Methods-Induced Biases at Subgroup Detection for Machine-Learning Based Data-Analysis of Biomedical Data. International Journal of Molecular Sciences, 21(1), 79. https://doi.org/10.3390/ijms21010079