
Evaluation of Stable LifeAct-mRuby2- and LAMP1-NeonGreen Expressing A549 Cell Lines for Investigation of Aspergillus fumigatus Interaction with Pulmonary Cells

 , ,
, , 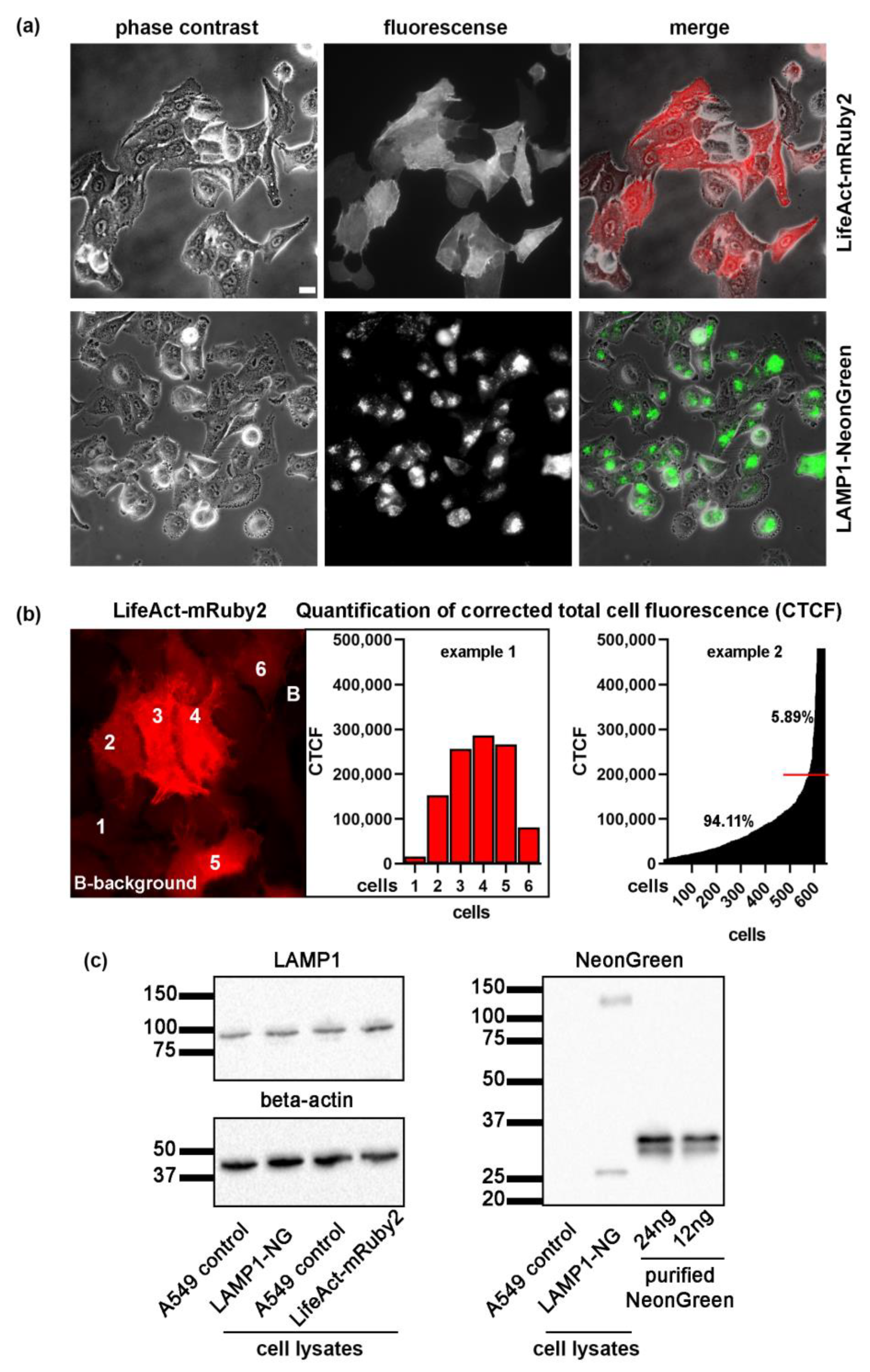{kind=link}
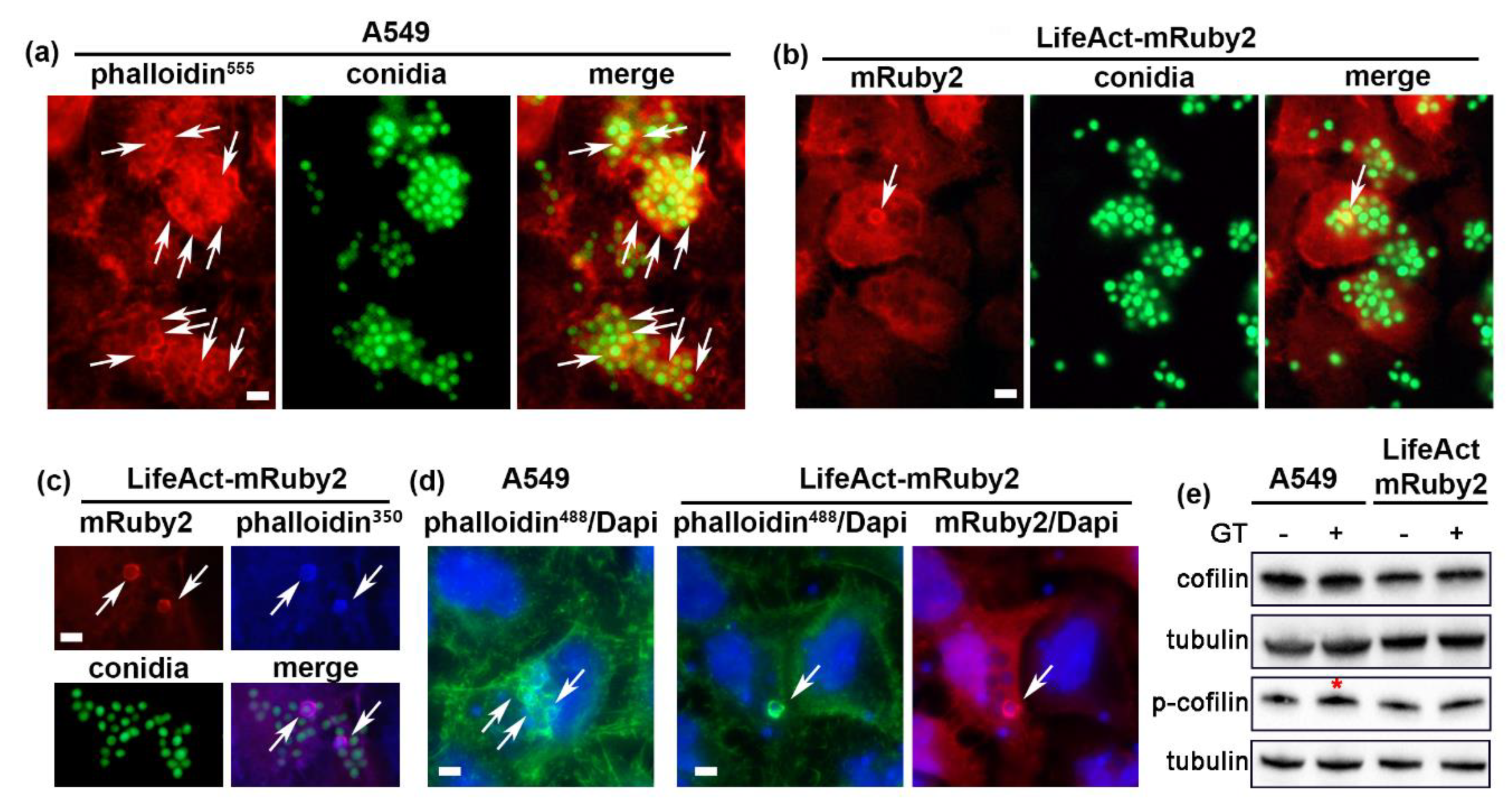{kind=link}
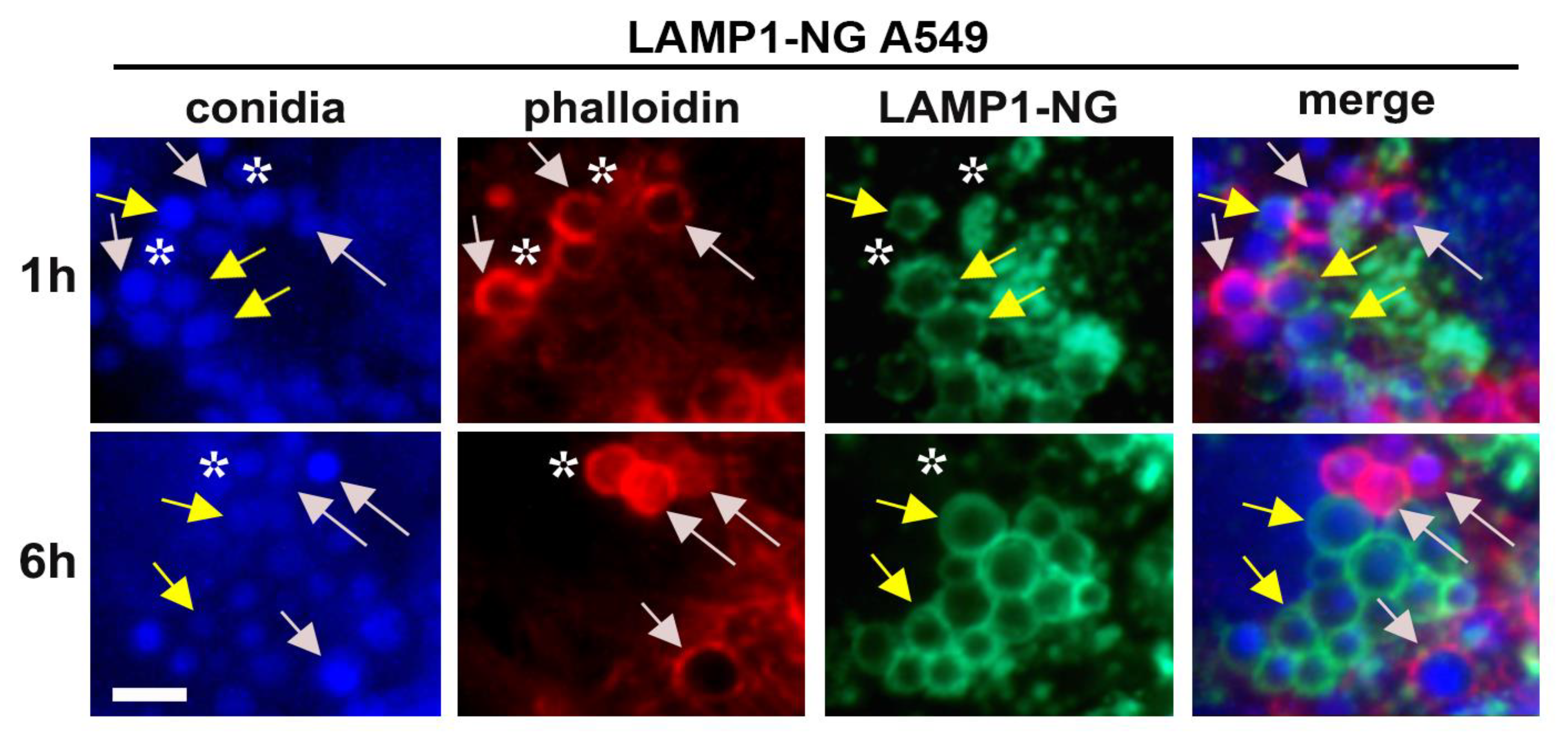{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Generation of A549 Cells Lines Stably Expressing LifeAct-mRuby2 and LAMP1
2.2. A. fumigatus Conidia Internalization in A549 Cells Stably Expressing LifeAct-mRuby2
2.3. A. fumigatus Conidia Internalization in A549 Cells Stably Expressing LAMP1
3. Materials and Methods
3.1. Reagents, Cell Culture, and Generation of Cell Lines
3.2. Preparation and Application of A. fumigatus Conidia
3.3. Immunofluorescence
3.4. Conidia Quantification
3.5. Western Blotting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wasylnka, J.A.; Moore, M.M. Uptake of Aspergillus fumigatus Conidia by phagocytic and nonphagocytic cells in vitro: Quantitation using strains expressing green fluorescent protein. Infect. Immun. 2002, 70, 3156–3163. [Google Scholar] [CrossRef] [Green Version]
- Paris, S.; Boisvieux-Ulrich, E.; Crestani, B.; Houcine, O.; Taramelli, D.; Lombardi, L.; Latgé, J.P. Internalization of Aspergillus fumigatus conidia by epithelial and endothelial cells. Infect. Immun. 1997, 65, 1510–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasylnka, J.A.; Moore, M.M. Aspergillus fumigatus conidia survive and germinate in acidic organelles of A549 epithelial cells. J. Cell Sci. 2003, 116, 1579–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botterel, F.; Gross, K.; Ibrahim-Granet, O.; Khoufache, K.; Escabasse, V.; Coste, A.; Cordonnier, C.; Escudier, E.; Bretagne, S. Phagocytosis of Aspergillus fumigatus conidia by primary nasal epithelial cells in vitro. BMC Microbiol. 2008, 8, 97. [Google Scholar] [CrossRef] [Green Version]
- Croft, C.A.; Culibrk, L.; Moore, M.M.; Tebbutt, S.J. Interactions of Aspergillus fumigatus Conidia with Airway Epithelial Cells: A Critical Review. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, P.; Hackett, T.L.; Moore, M.M.; Knight, D.A.; Tebbutt, S.J. Functional genomics of human bronchial epithelial cells directly interacting with conidia of Aspergillus fumigatus. BMC Gemonics 2010, 11, 358. [Google Scholar] [CrossRef] [Green Version]
- Aderem, A.; Underhill, D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999, 17, 593–623. [Google Scholar] [CrossRef]
- Ibrahim-Granet, O.; Philippe, B.; Boleti, H.; Boisvieux-Ulrich, E.; Grenet, D.; Stern, M.; Latgé, J.P. Phagocytosis and intracellular fate of Aspergillus fumigatus conidia in alveolar macrophages. Infect. Immun. 2003, 71, 891–903. [Google Scholar] [CrossRef] [Green Version]
- Stappers, M.H.T.; Clark, A.E.; Aimanianda, V.; Bidula, S.; Reid, D.M.; Asamaphan, P.; Hardison, S.E.; Dambuza, I.M.; Valsecchi, I.; Kerscher, B.; et al. Recognition of DHN-melanin by a C-type lectin receptor is required for immunity to Aspergillus. Nature 2018, 555, 382–386. [Google Scholar] [CrossRef]
- Greenberg, S. Modular components of phagocytosis. J. Leukoc. Biol. 1999, 66, 712–717. [Google Scholar] [CrossRef] [Green Version]
- Tjelle, T.E.; Lovdal, T.; Berg, T. Phagosome dynamics and function. BioEssays 2000, 22, 255–263. [Google Scholar] [CrossRef]
- Zheng, K.; Kitazato, K.; Wang, Y.; He, Z. Pathogenic microbes manipulate cofilin activity to subvert actin cytoskeleton. Crit. Rev. Microbiol. 2016, 42, 677–695. [Google Scholar] [CrossRef]
- Culibrk, L.; Croft, C.A.; Toor, A.; Yang, S.J.; Singhera, G.K.; Dorscheid, D.R.; Moore, M.M.; Tebbutt, S.J. Phagocytosis of Aspergillus fumigatus by Human Bronchial Epithelial Cells Is Mediated by the Arp2/3 Complex and WIPF2. Front. Cell. Infect. Microbiol. 2019, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Han, X.; Chen, F.; Jia, X.; Zhao, J.; Zhang, C.; Yong, C.; Tian, S.; Zhou, X.; Han, L. Evidence for the involvement of cofilin in Aspergillus fumigatus internalization into type II alveolar epithelial cells. BMC Microbiol. 2015, 15, 161. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Chen, F.; Liu, X.; Han, X.; Hu, Y.; Su, X.; Chen, Y.; Sun, Y.; Han, L. Gliotoxin Induces Cofilin Phosphorylation to Promote Actin Cytoskeleton Dynamics and Internalization of Aspergillus fumigatus Into Type II Human Pneumocyte Cells. Front. Microbiol. 2019, 10, 1345. [Google Scholar] [CrossRef] [PubMed]
- Melak, M.; Plessner, M.; Grosse, R. Actin visualization at a glance. J. Cell Sci. 2017, 130, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Courtemanche, N.; Pollard, T.D.; Chen, Q. Avoiding artefacts when counting polymerized actin in live cells with LifeAct fused to fluorescent proteins. Nat. Cell Biol. 2016, 18, 676–683. [Google Scholar] [CrossRef] [Green Version]
- Asakura, T.; Sasaki, T.; Nagano, F.; Satoh, A.; Obaishi, H.; Nishioka, H.; Imamura, H.; Hotta, K.; Tanaka, K.; Nakanishi, H.; et al. Isolation and characterization of a novel actin filament-binding protein from Saccharomyces cerevisiae. Oncogene 1998, 16, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedl, J.; Crevenna, A.H.; Kessenbrock, K.; Yu, J.H.; Neukirchen, D.; Bista, M.; Bradke, F.; Jenne, D.; Holak, T.A.; Werb, Z.; et al. Lifeact: A versatile marker to visualize F-actin. Nat. Methods 2008, 5, 605–607. [Google Scholar] [CrossRef]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Yordanov, T.E.; Hipolito, V.E.B.; Liebscher, G.; Vogel, G.F.; Stasyk, T.; Herrmann, C.; Geley, S.; Teis, D.; Botelho, R.J.; Hess, M.W.; et al. Biogenesis of lysosome-related organelles complex-1 (BORC) regulates late endosomal/lysosomal size through PIKfyve-dependent phosphatidylinositol-3,5-bisphosphate. Traffic 2019, 20, 674–696. [Google Scholar] [CrossRef] [Green Version]
- Hammond, L. Measuring Cell Fluorescence Using ImageJ. 2014. Available online: https://theolb.readthedocs.io/en/latest/imaging/measuring-cell-fluorescence-using-imagej.html (accessed on 25 April 2021).
- Burgess, A.; Vigneron, S.; Brioudes, E.; Labbé, J.-C.; Lorca, T.; Castro, A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc. Natl. Acad. Sci. USA 2010, 107, 12564–12569. [Google Scholar] [CrossRef] [Green Version]
- Gavet, O.; Pines, J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 2010, 18, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Munsie, L.N.; Caron, N.; Desmond, C.R.; Truant, R. Lifeact cannot visualize some forms of stress-induced twisted F-actin. Nat. Methods 2009, 6, 317. [Google Scholar] [CrossRef]
- Flores, L.R.; Keeling, M.C.; Zhang, X.; Sliogeryte, K.; Gavara, N. Lifeact-GFP alters F-actin organization, cellular morphology and biophysical behaviour. Sci. Rep. 2019, 9, 3241. [Google Scholar] [CrossRef]
- Yamashiro, S.; Taniguchi, D.; Tanaka, S.; Kiuchi, T.; Vavylonis, D.; Watanabe, N. Convection-Induced Biased Distribution of Actin Probes in Live Cells. Biophys. J. 2019, 116, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Schiefermeier-Mach, N.; Perkhofer, S.; Heinrich, L.; Haller, T. Stimulation of surfactant exocytosis in primary alveolar type II cells by A. fumigatus. Med. Mycol. 2020. [Google Scholar] [CrossRef]
- Scheffler, J.M.; Schiefermeier, N.; Huber, L.A. Mild fixation and permeabilization protocol for preserving structures of endosomes, focal adhesions, and actin filaments during immunofluorescence analysis. Meth. Enzymol. 2014, 535, 93–102. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Schiefermeier, N.; Scheffler, J.M.; de Araujo, M.E.G.; Stasyk, T.; Yordanov, T.; Ebner, H.L.; Offterdinger, M.; Munck, S.; Hess, M.W.; Wickström, S.A.; et al. The late endosomal p14-MP1 (LAMTOR2/3) complex regulates focal adhesion dynamics during cell migration. J. Cell Biol. 2014, 205, 525–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiefermeier-Mach, N.; Moresco, V.; Geley, S.; Heinrich, L.; Lechner, L.; Oberhauser, H.; Perkhofer, S. Evaluation of Stable LifeAct-mRuby2- and LAMP1-NeonGreen Expressing A549 Cell Lines for Investigation of Aspergillus fumigatus Interaction with Pulmonary Cells. Int. J. Mol. Sci. 2021, 22, 5965. https://doi.org/10.3390/ijms22115965
Schiefermeier-Mach N, Moresco V, Geley S, Heinrich L, Lechner L, Oberhauser H, Perkhofer S. Evaluation of Stable LifeAct-mRuby2- and LAMP1-NeonGreen Expressing A549 Cell Lines for Investigation of Aspergillus fumigatus Interaction with Pulmonary Cells. International Journal of Molecular Sciences. 2021; 22(11):5965. https://doi.org/10.3390/ijms22115965
Chicago/Turabian StyleSchiefermeier-Mach, Natalia, Violetta Moresco, Stephan Geley, Lea Heinrich, Lukas Lechner, Heidi Oberhauser, and Susanne Perkhofer. 2021. "Evaluation of Stable LifeAct-mRuby2- and LAMP1-NeonGreen Expressing A549 Cell Lines for Investigation of Aspergillus fumigatus Interaction with Pulmonary Cells" International Journal of Molecular Sciences 22, no. 11: 5965. https://doi.org/10.3390/ijms22115965
APA StyleSchiefermeier-Mach, N., Moresco, V., Geley, S., Heinrich, L., Lechner, L., Oberhauser, H., & Perkhofer, S. (2021). Evaluation of Stable LifeAct-mRuby2- and LAMP1-NeonGreen Expressing A549 Cell Lines for Investigation of Aspergillus fumigatus Interaction with Pulmonary Cells. International Journal of Molecular Sciences, 22(11), 5965. https://doi.org/10.3390/ijms22115965