
Glucocorticoid Signaling and Epigenetic Alterations in Stress-Related Disorders



Abstract
:1. Introduction
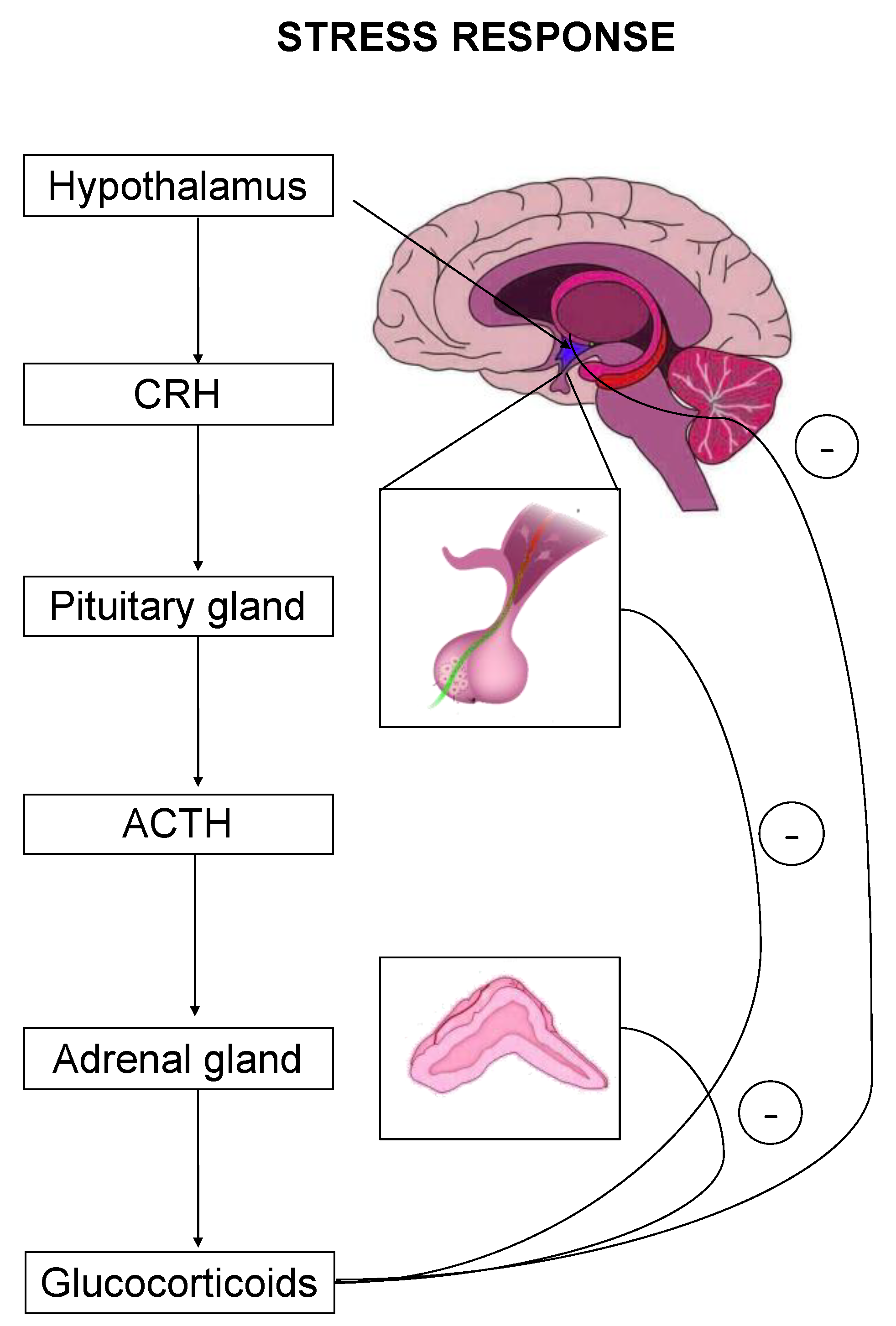
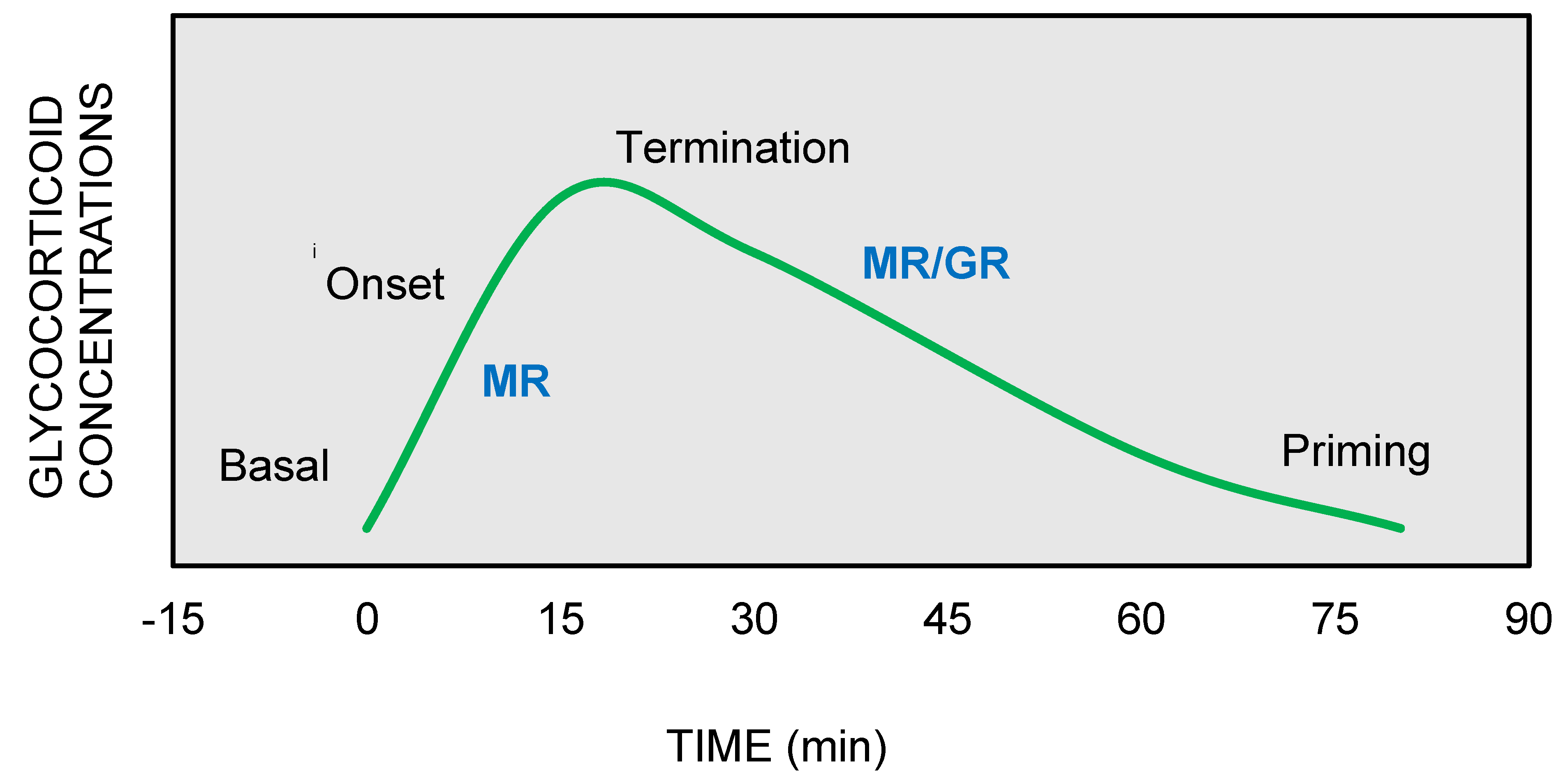
2. Glucocorticoid Signaling via GRs and MRs upon Stress Response
3. Functional Synergistic Network of MR and GR
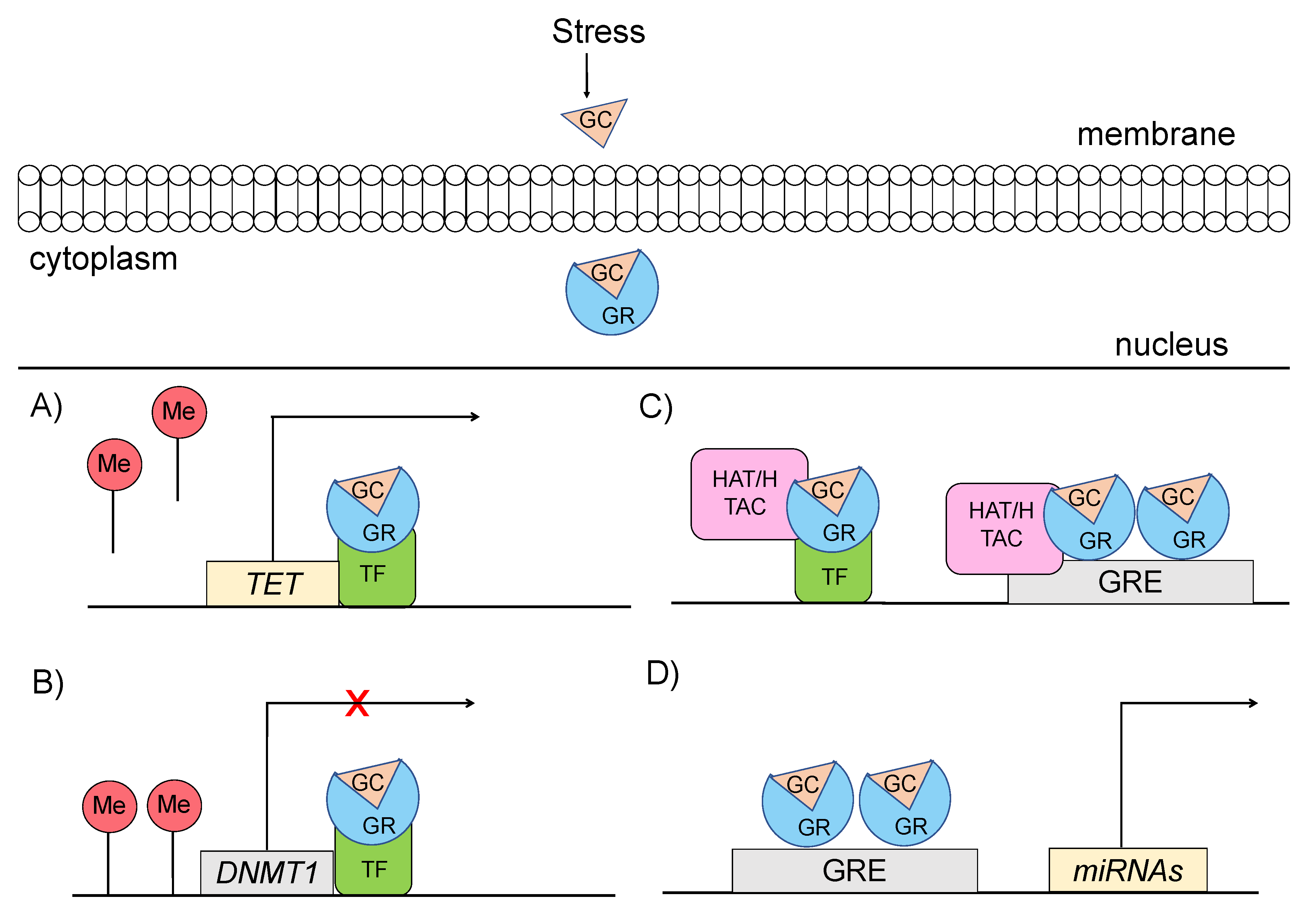
4. Epigenetic Alterations as a Cause of Dysregulation of the Stress Response
5. Epigenetic Changes and Stress-Related Disorders
6. Epigenetic Effects of Stress Exposure: Challenges and Confounders
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Charmandari, E.; Tsigos, C.; Chrousos, G. Endocrinology of the stress response. Annu. Rev. Physiol. 2005, 67, 259–284. [Google Scholar] [CrossRef]
- Chrousos, G.P. Stress and disorders of the stress system. Nat. Rev. Endocrinol. 2009, 5, 374–381. [Google Scholar] [CrossRef]
- Zannas, A.S.; Chrousos, G.P. Epigenetic programming by stress and glucocorticoids along the human lifespan. Mol. Psychiatry 2017, 22, 640–646. [Google Scholar] [CrossRef]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, Mechanisms and Clinical Perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Habib, K.E.; Gold, P.W.; Chrousos, G.P. Neuroendocrinology of stress. Endocrinol. Metab. Clin. N. Am. 2001, 30, 695–728. [Google Scholar] [CrossRef]
- Chrousos, G.P. Regulation and dysregulation of the hypothalamic-pituitary-adrenal axis. The corticotropin-releasing hormone perspective. Endocrinol. Metab. Clin. N. Am. 1992, 21, 833–858. [Google Scholar] [CrossRef]
- Whitnall, M.H. Regulation of the hypothalamic corticotropin-releasing hormone neurosecretory system. Prog. Neurobiol. 1993, 40, 573–629. [Google Scholar] [CrossRef]
- Nicolaides, N.C.; Kyratzi, E.; Lamprokostopoulou, A.; Chrousos, G.P.; Charmandari, E. Stress, the stress system and the role of glucocorticoids. Neuroimmunomodulation 2015, 22, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Ballard, P.L.; Baxter, J.D.; Higgins, S.J.; Rousseau, G.G.; Tomkins, G.M. General Presence of Glucocorticoid Receptors in Mammalian Tissues1. Endocrinology 1974, 94, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, N.C.; Charmandari, E.; Chrousos, G.P.; Kino, T. Recent advances in the molecular mechanisms determining tissue sensitivity to glucocorticoids: Novel mutations, circadian rhythm and ligand-induced repression of the human glucocorticoid receptor. BMC Endocr. Disord. 2014, 14, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, R. Molecular mechanisms of glucocorticoid action: What is important? Thorax 2000, 55, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Groeneweg, F.L.; Karst, H.; de Kloet, E.R.; Joëls, M. Rapid Non-Genomic Effects of Corticosteroids and Their Role in the Central Stress Response. J. Endocrinol. 2011, 209, pp. 153–167. Available online: https://joe.bioscientifica.com/view/journals/joe/209/2/153.xml (accessed on 15 March 2021).
- Mechanisms of the Anti-Inflammatory Effects of Glucocorticoids: Genomic and Nongenomic Interference with MAPK Signaling Pathways—Aναζήτηση Google. Available online: https://www.google.com/search?q=Mechanisms+of+the+anti-inflammatory+effects+of+glucocorticoids%3A+genomic+and+nongenomic+interference+with+MAPK+signaling+pathways&rlz=1C5CHFA_enGR909GR909&oq=Mechanisms+of+the+anti-inflammatory+effects+of+glucocorticoids%3A+genomic+and+nongenomic+interference+with+MAPK+signaling+pathways&aqs=chrome..69i57.801j0j7&sourceid=chrome&ie=UTF-8 (accessed on 15 March 2021).
- Song, I.-H.; Buttgereit, F. Non-genomic glucocorticoid effects to provide the basis for new drug developments. Mol. Cell Endocrinol. 2006, 246, 142–146. [Google Scholar] [CrossRef]
- Nicolaides, N.C.; Kino, T.; Chrousos, G.; Charmandari, E. Primary Generalized Glucocorticoid Resistance or Chrousos Syndrome. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: http://www.ncbi.nlm.nih.gov/books/NBK278930/ (accessed on 15 March 2021).
- Buttgereit, F.; Scheffold, A. Rapid glucocorticoid effects on immune cells. Steroids 2002, 67, 529–534. [Google Scholar] [CrossRef]
- Reul, J.M.; de Kloet, E.R. Two receptor systems for corticosterone in rat brain: Microdistribution and differential occupation. Endocrinology 1985, 117, 2505–2511. [Google Scholar] [CrossRef]
- Grossmann, C.; Scholz, T.; Rochel, M.; Bumket-Vogt, C.; Oelkers, W.; Pfeiffer, A.F.H.; Diederich, S.; Bahr, V. Transactivation via the human glucocorticoid and mineralocorticoid receptor by therapeutically used steroids in CV-1 cells: A comparison of their glucocorticoid and mineralocorticoid properties. Eur. J. Endocrinol. 2004, 151, 397–406. [Google Scholar] [CrossRef]
- Ter Heegde, F.; De Rijk, R.H.; Vinkers, C.H. The brain mineralocorticoid receptor and stress resilience. Psychoneuroendocrinology 2015, 52, 92–110. [Google Scholar] [CrossRef] [Green Version]
- Latouche, C.; Sainte-Marie, Y.; Steenman, M.; Castro Chaves, P.; Naray-Fejes-Toth, A.; Fejes-Toth, G.; Farman, N.; Jaisser, F. Molecular signature of mineralocorticoid receptor signaling in cardiomyocytes: From cultured cells to mouse heart. Endocrinology 2010, 151, 4467–4476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soundararajan, R.; Zhang, T.T.; Wang, J.; Vandewalle, A.; Pearce, D. A novel role for glucocorticoid-induced leucine zipper protein in epithelial sodium channel-mediated sodium transport. J. Biol. Chem. 2005, 280, 39970–39981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mifsud, K.R.; Reul, J.M.H.M. Acute stress enhances heterodimerization and binding of corticosteroid receptors at glucocorticoid target genes in the hippocampus. Proc. Natl. Acad. Sci. USA 2016, 113, 11336–11341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Bhargava, A.; Mastroberardino, L.; Meijer, O.C.; Wang, J.; Buse, P.; Firestone, G.L.; Verrey, F.; Pearce, D. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc. Natl. Acad. Sci. USA 1999, 96, 2514–2519. [Google Scholar] [CrossRef] [Green Version]
- Groeneweg, F.L.; Karst, H.; de Kloet, E.R.; Joëls, M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol. Cell Endocrinol. 2012, 350, 299–309. [Google Scholar] [CrossRef]
- Karst, H.; Berger, S.; Turiault, M.; Tronche, F.; Schütz, G.; Joëls, M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Natl. Acad. Sci. USA 2005, 102, 19204–19207. [Google Scholar] [CrossRef] [Green Version]
- Karst, H.; Berger, S.; Erdmann, G.; Schütz, G.; Joëls, M. Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc. Natl. Acad. Sci. USA 2010, 107, 14449–14454. [Google Scholar] [CrossRef] [Green Version]
- MR/GR Signaling in the Brain during the Stress Response|IntechOpen. Available online: https://www.intechopen.com/books/aldosterone-mineralocorticoid-receptor-cell-biology-to-translational-medicine/mr-gr-signaling-in-the-brain-during-the-stress-response (accessed on 17 March 2021).
- Joëls, M.; Karst, H.; DeRijk, R.; de Kloet, E.R. The coming out of the brain mineralocorticoid receptor. Trends Neurosci. 2008, 31, 1–7. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Joëls, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef]
- De Kloet, E.R.; Meijer, O.C.; de Nicola, A.F.; de Rijk, R.H.; Joëls, M. Importance of the brain corticosteroid receptor balance in metaplasticity, cognitive performance and neuro-inflammation. Front. Neuroendocrinol. 2018, 49, 124–145. [Google Scholar] [CrossRef]
- Sarabdjitsingh, R.A.; Jezequel, J.; Pasricha, N.; Mikasova, L.; Kerkhofs, A.; Karst, H.; Groc, L.; Joels, M. Ultradian corticosterone pulses balance glutamatergic transmission and synaptic plasticity. Proc. Natl. Acad. Sci. USA 2014, 111, 14265–14270. [Google Scholar] [CrossRef] [Green Version]
- De Kloet, E.R.; Reul, J.M. Feedback action and tonic influence of corticosteroids on brain function: A concept arising from the heterogeneity of brain receptor systems. Psychoneuroendocrinology 1987, 12, 83–105. [Google Scholar] [CrossRef]
- Cornelisse, S.; Joëls, M.; Smeets, T. A Randomized Trial on Mineralocorticoid Receptor Blockade in Men: Effects on Stress Responses, Selective Attention, and Memory. Neuropsychopharmacology 2011, 36, 2720–2728. [Google Scholar] [CrossRef] [Green Version]
- Korte, S.M.; De Boer, S.F.; De Kloet, E.R.; Bohus, B. Anxiolytic-like effects of selective mineralocorticoid and glucocorticoid antagonists on fear-enhanced behavior in the elevated plus-maze. Psychoneuroendocrinology 1995, 20, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Kruk, M.R.; Haller, J.; Meelis, W.; de Kloet, E.R. Mineralocorticoid receptor blockade during a rat’s first violent encounter inhibits its subsequent propensity for violence. Behav. Neurosci. 2013, 127, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Kloet, E.R.; Vreugdenhil, E.; Oitzl, M.S.; Joëls, M. Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 1998, 19, 269–301. [Google Scholar] [CrossRef]
- Russell, G.M.; Henley, D.E.; Leendertz, J.; Douthwaite, J.A.; Wood, S.A.; Stevens, A.; Woltersdorf, W.W.; Peeters, B.W.M.M.; Ruigt, G.S.F.; White, A.; et al. Rapid glucocorticoid receptor-mediated inhibition of hypothalamic-pituitary-adrenal ultradian activity in healthy males. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 6106–6115. [Google Scholar] [CrossRef] [Green Version]
- Keller-Wood, M.E.; Dallman, M.F. Corticosteroid inhibition of ACTH secretion. Endocr. Rev. 1984, 5, 1–24. [Google Scholar] [CrossRef]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the hypothalamic-pituitary-adrenocortical stress response. Compr. Physiol. 2016, 6, 603–621. [Google Scholar] [CrossRef] [Green Version]
- Oitzl, M.S.; Reichardt, H.M.; Joëls, M.; de Kloet, E.R. Point mutation in the mouse glucocorticoid receptor preventing DNA binding impairs spatial memory. Proc. Natl. Acad. Sci. USA 2001, 98, 12790–12795. [Google Scholar] [CrossRef] [Green Version]
- Henckens, M.J.A.G.; Pu, Z.; Hermans, E.J.; van Wingen, G.A.; Joëls, M.; Fernández, G. Dynamically changing effects of corticosteroids on human hippocampal and prefrontal processing. Hum. Brain Mapp. 2011, 33, 2885–2897. [Google Scholar] [CrossRef]
- Feng, X.; Zhao, Y.; Yang, T.; Song, M.; Wang, C.; Yao, Y.; Fan, H. Glucocorticoid-Driven NLRP3 Inflammasome Activation in Hippocampal Microglia Mediates Chronic Stress-Induced Depressive-Like Behaviors. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef]
- Frank, M.G.; Hershman, S.A.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Chronic exposure to exogenous glucocorticoids primes microglia to pro-inflammatory stimuli and induces NLRP3 mRNA in the hippocampus. Psychoneuroendocrinology 2014, 40, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.G.; Watkins, L.R.; Maier, S.F. The permissive role of glucocorticoids in neuroinflammatory priming: Mechanisms and insights. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 300–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Kloet, E.R.; Meijer, O.C. MR/GR Signaling in the Brain during the Stress Response. Aldosterone-Miner. Recept Cell Biol. Transl. Med. 2019. [Google Scholar] [CrossRef] [Green Version]
- Gassen, N.C.; Chrousos, G.P.; Binder, E.B.; Zannas, A.S. Life stress, glucocorticoid signaling, and the aging epigenome: Implications for aging-related diseases. Neurosci. Biobehav. Rev. 2017, 74 Pt B, 356–365. [Google Scholar] [CrossRef]
- Herceg, Z. Epigenetic Mechanisms as an Interface Between the Environment and Genome. In Hypoxia: Translation in Progress; Roach, R.C., Hackett, P.H., Wagner, P.D., Eds.; Advances in Experimental Medicine and Biology; Springer US: New York, NY, USA, 2016; pp. 3–15. [Google Scholar] [CrossRef]
- Bagot, R.C.; Labonté, B.; Peña, C.J.; Nestler, E.J. Epigenetic signaling in psychiatric disorders: Stress and depression. Dialogues Clin. Neurosci. 2014, 16, 281–295. [Google Scholar]
- Alyamani, R.A.S.; Murgatroyd, C. Epigenetic Programming by Early-Life Stress. Prog. Mol. Biol. Transl. Sci. 2018, 157, 133–150. [Google Scholar] [CrossRef]
- Wiechmann, T.; Röh, S.; Sauer, S.; Czamara, D.; Arloth, J.; Ködel, M.; Beintner, M.; Knop, L.; Menke, A.; Binder, E.B.; et al. Identification of dynamic glucocorticoid-induced methylation changes at the FKBP5 locus. Clin. Epigenet. 2019, 11, 83. [Google Scholar] [CrossRef]
- Park, C.; Rosenblat, J.D.; Brietzke, E.; Pan, Z.; Lee, Y.; Cao, B.; Zuckerman, H.; Kalantarova, A.; McIntyre, R.S. Stress, epigenetics and depression: A systematic review. Neurosci. Biobehav. Rev. 2019, 102, 139–152. [Google Scholar] [CrossRef]
- Uht, R.M. Mechanisms of Glucocorticoid Receptor (GR) Mediated Corticotropin Releasing Hormone Gene Expression. Glucocorticoids New Recognit. Fam. Friend 2012. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, Y.; Roy, B.; Lugli, G.; Rizavi, H.; Zhang, H.; Smalheiser, N.R. Chronic corticosterone-mediated dysregulation of microRNA network in prefrontal cortex of rats: Relevance to depression pathophysiology. Transl. Psychiatry 2015, 5, e682. [Google Scholar] [CrossRef] [PubMed]
- Vockley, C.M.; D’Ippolito, A.M.; McDowell, I.C.; Majoros, W.H.; Safi, A.; Song, L.; Crawford, G.E.; Reddy, T.E. Direct GR binding sites potentiate clusters of TF binding across the human genome. Cell 2016, 166, 1269–1281.e19. [Google Scholar] [CrossRef] [Green Version]
- Non, A.L.; Binder, A.M.; Kubzansky, L.D.; Michels, K.B. Genome-wide DNA methylation in neonates exposed to maternal depression, anxiety, or SSRI medication during pregnancy. Epigenetics 2014, 9, 964–972. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Postovit, L.; Cattaneo, A.; Binder, E.B.; Aitchison, K.J. Epigenetic Modifications in Stress Response Genes Associated With Childhood Trauma. Front. Psychiatry 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiele, M.A.; Gottschalk, M.G.; Domschke, K. The applied implications of epigenetics in anxiety, affective and stress-related disorders—A review and synthesis on psychosocial stress, psychotherapy and prevention. Clin. Psychol. Rev. 2020, 77, 101830. [Google Scholar] [CrossRef]
- FKBP5 Allele-Specific Epigenetic Modification in Gene by Environment Interaction-PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/25482174/ (accessed on 26 March 2021).
- Zannas, A.S.; Wiechmann, T.; Gassen, N.C.; Binder, E.B. Gene–Stress–Epigenetic Regulation of FKBP5: Clinical and Translational Implications. Neuropsychopharmacology 2016, 41, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Scharf, S.H.; Liebl, C.; Binder, E.B.; Schmidt, M.V.; Müller, M.B. Expression and Regulation of the Fkbp5 Gene in the Adult Mouse Brain. PLoS ONE 2011, 6, e16883. [Google Scholar] [CrossRef] [Green Version]
- van der Knaap, L.J.; Riese, H.; Hudziak, J.J.; Verbiest, M.M.P.J.; Verhulst, F.C.; Oldehinkel, A.J. Glucocorticoid receptor gene (NR3C1) methylation following stressful events between birth and adolescence. The TRAILS study. Transl. Psychiatry 2014, 4, e381. [Google Scholar] [CrossRef]
- Vukojevic, V.; Kolassa, I.-T.; Fastenrath, M.; Gschwind, L.; Spalek, K.; Milnik, A.; Heck, A.; Vogler, C.; Wilker, S.; Demougin, P.; et al. Epigenetic Modification of the Glucocorticoid Receptor Gene Is Linked to Traumatic Memory and Post-Traumatic Stress Disorder Risk in Genocide Survivors. J. Neurosci. 2014, 34, 10274–10284. [Google Scholar] [CrossRef]
- Jimeno, B.; Hau, M.; Gómez-Díaz, E.; Verhulst, S. Developmental conditions modulate DNA methylation at the glucocorticoid receptor gene with cascading effects on expression and corticosterone levels in zebra finches. Sci. Rep. 2019, 9, 15869. [Google Scholar] [CrossRef]
- Ferrer, A.; Labad, J.; Salvat-Pujol, N.; Barrachina, M.; Costas, J.; Urretavizcaya, M.; de Arriba-Arnau, A.; Crespo, J.M.; Soriano-Mas, C.; Carracedo, Á.; et al. BDNF genetic variants and methylation: Effects on cognition in major depressive disorder. Transl. Psychiatry 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Fachim, H.A.; Corsi-Zuelli, F.; Loureiro, C.M.; Iamjan, S.-A.; Shuhama, R.; Joca, S.; Menezes, P.R.; Heald, A.; Louzada-Junior, P.; Dalton, C.F.; et al. Early-Life Stress Effects on BDNF DNA Methylation in First-Episode Psychosis and in Rats Reared in Isolation. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 108, 110188. [Google Scholar] [CrossRef]
- Roth, T.L.; Zoladz, P.R.; Sweatt, J.D.; Diamond, D.M. Epigenetic Modification of Hippocampal Bdnf DNA in Adult Rats in an Animal Model of Post-Traumatic Stress Disorder. J. Psychiatr. Res. 2011, 45, 919–926. [Google Scholar] [CrossRef] [Green Version]
- Voisey, J.; Lawford, B.; Bruenig, D.; Harvey, W.; Morris, C.P.; Young, R.M.; Mehta, D.; PTSD Initiative. Differential BDNF methylation in combat exposed veterans and the association with exercise. Gene 2019, 698, 107–112. [Google Scholar] [CrossRef]
- Fuchikami, M.; Yamamoto, S.; Morinobu, S.; Takei, S.; Yamawaki, S. Epigenetic Regulation of BDNF Gene in Response to Stress. Psychiatry Investig. 2010, 7, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Provenzi, L.; Giorda, R.; Beri, S.; Montirosso, R. SLC6A4 methylation as an epigenetic marker of life adversity exposures in humans: A systematic review of literature. Neurosci. Biobehav. Rev. 2016, 71, 7–20. [Google Scholar] [CrossRef]
- Kang, H.-J.; Kim, J.-M.; Stewart, R.; Kim, S.-Y.; Bae, K.-Y.; Kim, S.-W.; Shin, I.-S.; Shin, M.-G.; Yoon, J.-S. Association of SLC6A4 methylation with early adversity, characteristics and outcomes in depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 44, 23–28. [Google Scholar] [CrossRef]
- Booij, L.; Szyf, M.; Carballedo, A.; Frey, E.-M.; Morris, D.; Dymov, S.; Vaisheva, F.; Ly, V.; Fahey, C.; Meaney, J.; et al. DNA methylation of the serotonin transporter gene in peripheral cells and stress-related changes in hippocampal volume: A study in depressed patients and healthy controls. PLoS ONE 2015, 10, e0119061. [Google Scholar] [CrossRef]
- van der Knaap, L.J.; Riese, H.; Hudziak, J.J.; Verbiest, M.M.P.J.; Verhulst, F.C.; Oldehinkel, A.J.; van Oort, F.V.A. Adverse life events and allele-specific methylation of the serotonin transporter gene (SLC6A4) in adolescents: The TRAILS study. Psychosom. Med. 2015, 77, 246–255. [Google Scholar] [CrossRef]
- Alasaari, J.S.; Lagus, M.; Ollila, H.M.; Toivola, A.; Kivimäki, M.; Vahtera, J.; Kronholm, E.; Härmä, M.; Puttonen, S.; Paunio, T. Environmental Stress Affects DNA Methylation of a CpG Rich Promoter Region of Serotonin Transporter Gene in a Nurse Cohort. PLoS ONE 2012, 7, e45813. [Google Scholar] [CrossRef]
- Wang, D.; Liu, X.; Zhou, Y.; Xie, H.; Hong, X.; Tsai, H.-J.; Wang, G.; Liu, R.; Wang, X. Individual variation and longitudinal pattern of genome-wide DNA methylation from birth to the first two years of life. Epigenetics 2012, 7, 594–605. [Google Scholar] [CrossRef] [Green Version]
- Duman, E.A.; Canli, T. Influence of life stress, 5-HTTLPR genotype, and SLC6A4 methylation on gene expression and stress response in healthy Caucasian males. Biol. Mood Anxiety Disord. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Kraaijenvanger, E.J.; He, Y.; Spencer, H.; Smith, A.K.; Bos, P.A.; Boks, M.P.M. Epigenetic Variability in the Human Oxytocin Receptor (OXTR) Gene: A Possible Pathway from Early Life Experiences to Psychopathologies. Neurosci. Biobehav. Rev. 2019, 96, 127–142. [Google Scholar] [CrossRef]
- Thaler, L.; Brassard, S.; Booij, L.; Kahan, E.; McGregor, K.; Labbe, A.; Israel, M.; Steiger, H. Methylation of the OXTR gene in women with anorexia nervosa: Relationship to social behavior. Eur. Eat. Disord. Rev. J. Eat. Disord. Assoc. 2020, 28, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Andari, E.; Nishitani, S.; Kaundinya, G.; Caceres, G.A.; Morrier, M.J.; Ousley, O.; Smith, A.K.; Cubells, J.F.; Young, L. Epigenetic modification of the oxytocin receptor gene: Implications for autism symptom severity and brain functional connectivity. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2020, 45, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Maud, C.; Ryan, J.; McIntosh, J.E.; Olsson, C.A. The role of oxytocin receptor gene (OXTR) DNA methylation (DNAm) in human social and emotional functioning: A systematic narrative review. BMC Psychiatry 2018, 18. [Google Scholar] [CrossRef] [Green Version]
- Kusui, C.; Kimura, T.; Ogita, K.; Nakamura, H.; Matsumura, Y.; Koyama, M.; Azuma, C.; Murata, Y. DNA methylation of the human oxytocin receptor gene promoter regulates tissue-specific gene suppression. Biochem. Biophys. Res. Commun. 2001, 289, 681–686. [Google Scholar] [CrossRef]
- Saito, T.; Shinozaki, G.; Koga, M.; Tanichi, M.; Takeshita, S.; Nakagawa, R.; Nagamine, M.; Cho, H.R.; Morimoto, Y.; Kobayashi, Y.; et al. Effect of interaction between a specific subtype of child abuse and the FKBP5 rs1360780 SNP on DNA methylation among patients with bipolar disorder. J. Affect. Disord. 2020, 272, 417–422. [Google Scholar] [CrossRef]
- Misiak, B.; Karpiński, P.; Szmida, E.; Grąźlewski, T.; Jabłoński, M.; Cyranka, K.; Rymaszewska, J.; Piotrowski, P.; Kotowicz, K.; Frydecka, D. Adverse Childhood Experiences and Methylation of the FKBP5 Gene in Patients with Psychotic Disorders. J. Clin. Med. 2020, 9, 3792. [Google Scholar] [CrossRef]
- Ramo-Fernández, L.; Boeck, C.; Koenig, A.M.; Schury, K.; Binder, E.B.; Gündel, H.; Fegert, J.M.; Karabatsiakis, A.; Kolassa, I.-T. The effects of childhood maltreatment on epigenetic regulation of stress-response associated genes: An intergenerational approach. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Borçoi, A.R.; Mendes, S.O.; Moreno, I.A.A.; Gasparini Dos Santos, J.; Freitas, F.V.; Pinheiro, J.A.; de Oliveira, M.M.; Barbosa, W.M.; Arpini, J.K.; Archanjo, A.B.; et al. Food and nutritional insecurity is associated with depressive symptoms mediated by NR3C1 gene promoter 1F methylation. Stress 2021, 1–8. [Google Scholar] [CrossRef]
- De Assis Pinheiro, J.; Freitas, F.V.; Borçoi, A.R.; Mendes, S.O.; Conti, C.L.; Arpini, J.K.; Dos Santos Vieira, T.; de Souza, R.A.; Dos Santos, D.P.; Barbosa, W.M.; et al. Alcohol consumption, depression, overweight and cortisol levels as determining factors for NR3C1 gene methylation. Sci. Rep. 2021, 11, 6768. [Google Scholar] [CrossRef]
- Misiak, B.; Samochowiec, J.; Konopka, A.; Gawrońska-Szklarz, B.; Beszłej, J.A.; Szmida, E.; Karpiński, P. Clinical correlates of the NR3C1 gene methylation at various stages of psychosis. Int. J. Neuropsychopharmacol. 2020, 24. [Google Scholar] [CrossRef]
- Duffy, H.B.D.; Roth, T.L. Increases in Bdnf DNA Methylation in the Prefrontal Cortex Following Aversive Caregiving Are Reflected in Blood Tissue. Front. Hum. Neurosci. 2020, 14, 594244. [Google Scholar] [CrossRef] [PubMed]
- Blaze, J.; Roth, T.L. Caregiver maltreatment causes altered neuronal DNA methylation in female rodents. Dev. Psychopathol. 2017, 29, 477–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niknazar, S.; Nahavandi, A.; Peyvandi, A.A.; Peyvandi, H.; Zare Mehrjerdi, F.; Karimi, M. Effect of Maternal Stress Prior to Conception on Hippocampal BDNF Signaling in Rat Offspring. Mol. Neurobiol. 2017, 54, 6436–6445. [Google Scholar] [CrossRef]
- DeLano, K.; Folger, A.T.; Ding, L.; Ji, H.; Yolton, K.; Ammerman, R.T.; Van Ginkel, J.B.; Bowers, K.A. Associations Between Maternal Community Deprivation and Infant DNA Methylation of the SLC6A4 Gene. Front. Public Health 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Timothy, A.; Benegal, V.; Shankarappa, B.; Saxena, S.; Jain, S.; Purushottam, M. Influence of early adversity on cortisol reactivity, SLC6A4 methylation and externalizing behavior in children of alcoholics. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 94. [Google Scholar] [CrossRef]
- Smith, J.A.; Zhao, W.; Wang, X.; Ratliff, S.M.; Mukherjee, B.; Kardia, S.L.R.; Liu, Y.; Roux, A.V.D.; Needham, B.L. Neighborhood characteristics influence DNA methylation of genes involved in stress response and inflammation: The Multi-Ethnic Study of Atherosclerosis. Epigenetics 2017, 12, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Kogan, S.M.; Bae, D.; Cho, J.; Smith, A.K.; Nishitani, S. Childhood Adversity, Socioeconomic Instability, Oxytocin-Receptor-Gene Methylation, and Romantic-Relationship Support Among Young African American Men. Psychol. Sci. 2019, 30, 1234–1244. [Google Scholar] [CrossRef]
- Kogan, S.M.; Cho, J.; Beach, S.R.H.; Smith, A.K.; Nishitani, S. Oxytocin receptor gene methylation and substance use problems among young African American men. Drug Alcohol Depend. 2018, 192, 309–315. [Google Scholar] [CrossRef]
- Gouin, J.P.; Zhou, Q.Q.; Booij, L.; Boivin, M.; Côté, S.M.; Hébert, M.; Ouellet-Morin, I.; Szyf, M.; Tremblay, R.E.; Turecki, G.; et al. Associations among oxytocin receptor gene (OXTR) DNA methylation in adulthood, exposure to early life adversity, and childhood trajectories of anxiousness. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Klengel, T.; Binder, E.B. Allele-specific epigenetic modification: A molecular mechanism for gene–environment interactions in stress-related psychiatric disorders? Epigenomics 2013, 5, 109–112. [Google Scholar] [CrossRef]
- Daskalakis, N.P.; Yehuda, R. Site-specific methylation changes in the glucocorticoid receptor exon 1F promoter in relation to life adversity: Systematic review of contributing factors. Front. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicchetti, D.; Handley, E.D. Methylation of the glucocorticoid receptor gene (NR3C1) in maltreated and nonmaltreated children: Associations with behavioral undercontrol, emotional lability/negativity, and externalizing and internalizing symptoms. Dev. Psychopathol. 2017, 29, 1795–1806. [Google Scholar] [CrossRef]
- Tyrka, A.R.; Parade, S.H.; Eslinger, N.M.; Marsit, C.J.; Lesseur, C.; Armstrong, D.A.; Philip, N.S.; Josefson, B.; Seifer, R. Methylation of Exons 1D, 1F, and 1H of the Glucocorticoid Receptor Gene Promoter and Exposure to Adversity in Pre-School Aged Children. Dev. Psychopathol. 2015, 27, 577–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma-Gudiel, H.; Córdova-Palomera, A.; Leza, J.C.; Fañanás, L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: A critical review. Neurosci. Biobehav. Rev. 2015, 55, 520–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onishchenko, N.; Karpova, N.; Sabri, F.; Castrén, E.; Ceccatelli, S. Long-lasting depression-like behavior and epigenetic changes of BDNF gene expression induced by perinatal exposure to methylmercury. J. Neurochem. 2008, 106, 1378–1387. [Google Scholar] [CrossRef]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef]
- Miao, Z.; Wang, Y.; Sun, Z. The Relationships Between Stress, Mental Disorders, and Epigenetic Regulation of BDNF. Int. J. Mol. Sci. 2020, 21, 1375. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, Y.; Rizavi, H.S.; Conley, R.R.; Roberts, R.C.; Tamminga, C.A.; Pandey, G.N. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch. Gen. Psychiatry 2003, 60, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Molendijk, M.; Bus, B.A.; Spinhoven, P.; Penninx, B.W.; Kenis, G.; Prickaerts, J.; Voshaar, R.O.; Elzinga, B. Serum levels of brain-derived neurotrophic factor in major depressive disorder: State-trait issues, clinical features and pharmacological treatment. Mol. Psychiatry 2011, 16, 1088–1095. [Google Scholar] [CrossRef] [Green Version]
- Gören, J.L. Brain-derived neurotrophic factor and schizophrenia. Ment. Health Clin. 2016, 6, 285–288. [Google Scholar] [CrossRef]
- Ursini, G.; Cavalleri, T.; Fazio, L.; Angrisano, T.; Iacovelli, L.; Porcelli, A.; Maddalena, G.; Punzi, G.; Mancini, M.; Gelao, B.; et al. BDNF rs6265 methylation and genotype interact on risk for schizophrenia. Epigenetics 2016, 11, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Mill, J.; Tang, T.; Kaminsky, Z.; Khare, T.; Yazdanpanah, S.; Bouchard, L.; Jia, P.; Assadzadeh, A.; Flanagan, J.; Schumacher, A.; et al. Epigenomic Profiling Reveals DNA-Methylation Changes Associated with Major Psychosis. Am. J. Hum. Genet. 2008, 82, 696–711. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.C.; Maffioletti, E.; Gennarelli, M.; Baune, B.T.; Minelli, A. Biological correlates of early life stressful events in major depressive disorder. Psychoneuroendocrinology 2021, 125, 105103. [Google Scholar] [CrossRef]
- Swartz, J.R.; Hariri, A.R.; Williamson, D.E. An epigenetic mechanism links socioeconomic status to changes in depression-related brain function in high-risk adolescents. Mol. Psychiatry 2017, 22, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Olsson, C.A.; Foley, D.L.; Parkinson-Bates, M.; Byrnes, G.; McKenzie, M.; Patton, G.C.; Morley, R.; Anney, R.J.L.; Craig, J.M.; Saffery, R. Prospects for epigenetic research within cohort studies of psychological disorder: A pilot investigation of a peripheral cell marker of epigenetic risk for depression. Biol. Psychol. 2010, 83, 159–165. [Google Scholar] [CrossRef]
- Lei, M.-K.; Beach, S.R.H.; Simons, R.L.; Philibert, R.A. Neighborhood crime and depressive symptoms among African American women: Genetic moderation and epigenetic mediation of effects. Soc. Sci. Med. 2015, 146, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Simons, R.L.; Lei, M.K.; Beach, S.R.H.; Cutrona, C.E.; Philibert, R.A. Methylation of the oxytocin receptor gene mediates the effect of adversity on negative schemas and depression. Dev. Psychopathol. 2017, 29, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Smearman, E.L.; Almli, L.M.; Conneely, K.N.; Brody, G.H.; Sales, J.M.; Bradley, B.; Ressler, K.J.; Smith, A.K. Oxytocin Receptor Genetic and Epigenetic Variations: Association With Child Abuse and Adult Psychiatric Symptoms. Child. Dev. 2016, 87, 122–134. [Google Scholar] [CrossRef] [Green Version]
- Mihaljevic, M.; Franic, D.; Soldatovic, I.; Lukic, I.; Petrovic, S.A.; Mirjanic, T.; Stankovic, B.; Zukic, B.; Zeljic, K.; Gasic, V.; et al. The FKBP5 genotype and childhood trauma effects on FKBP5 DNA methylation in patients with psychosis, their unaffected siblings, and healthy controls. Psychoneuroendocrinology 2021, 128, 105205. [Google Scholar] [CrossRef]
- Kang, J.I.; Kim, T.Y.; Choi, J.H.; So, H.S.; Kim, S.J. Allele-specific DNA methylation level of FKBP5 is associated with post-traumatic stress disorder. Psychoneuroendocrinology 2019, 103, 1–7. [Google Scholar] [CrossRef]
- Klinger-König, J.; Hertel, J.; Van der Auwera, S.; Frenzel, S.; Pfeiffer, L.; Waldenberger, M.; Golchert, J.; Teumer, A.; Nauck, M.; Homuth, G.; et al. Methylation of the FKBP5 gene in association with FKBP5 genotypes, childhood maltreatment and depression. Neuropsychopharmacology 2019, 44, 930–938. [Google Scholar] [CrossRef]
- Bakusic, J.; Vrieze, E.; Ghosh, M.; Bekaert, B.; Claes, S.; Godderis, L. Increased methylation of NR3C1 and SLC6A4 is associated with blunted cortisol reactivity to stress in major depression. Neurobiol. Stress 2020, 13, 100272. [Google Scholar] [CrossRef]
- Guo, J.; Yang, Y.; Jiang, X.; Guo, M.; Li, X.; Huang, P.; Liu, Z. Differential promoter methylation and G-712A polymorphism of brain-derived neurotrophic factor in post-traumatic stress disorder patients of Li and Han populations in Hainan province. Gene 2021, 769. [Google Scholar] [CrossRef]
- Hossack, M.R.; Reid, M.W.; Aden, J.K.; Gibbons, T.; Noe, J.C.; Willis, A.M. Adverse Childhood Experience, Genes, and PTSD Risk in Soldiers: A Methylation Study. In Military Medicine; Oxford University Press: Oxford, UK, 2020; Volume 185, pp. 377–384. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Zhu, Y.; Strachan, E.; Fowler, E.; Bacus, T.; Roy-Byrne, P.; Goldberg, J.; Vaccarino, V.; Zhao, J. Childhood Trauma, DNA Methylation of Stress-Related Genes, and Depression: Findings from Two Monozygotic Twin Studies. Psychosom. Med. 2018, 80, 599–608. [Google Scholar] [CrossRef]
- Shirata, T.; Suzuki, A.; Matsumoto, Y.; Noto, K.; Goto, K.; Otani, K. Interrelation between increased bdnf gene methylation and high sociotropy, a personality vulnerability factor in cognitive model of depression. Neuropsychiatr. Dis. Treat. 2020, 16, 1257–1263. [Google Scholar] [CrossRef]
- Sanwald, S.; Widenhorn-Müller, K.; Schönfeldt-Lecuona, C.; GenEmo Research Group; Montag, C.; Kiefer, M. Factors related to age at depression onset: The role of SLC6A4 methylation, sex, exposure to stressful life events and personality in a sample of inpatients suffering from major depression. BMC Psychiatry 2021, 21. [Google Scholar] [CrossRef]
- Schneider, I.; Kugel, H.; Redlich, R.; Grotegerd, D.; Bürger, C.; Bürkner, P.-C.; Opel, N.; Dohm, K.; Zaremba, D.; Meinert, S.; et al. Association of Serotonin Transporter Gene AluJb Methylation with Major Depression, Amygdala Responsiveness, 5-HTTLPR/rs25531 Polymorphism, and Stress. Neuropsychopharmacology 2018, 43, 1308–1316. [Google Scholar] [CrossRef]
- Nawijn, L.; Krzyzewska, I.M.; van Zuiden, M.; Henneman, P.; Koch, S.B.J.; Mul, A.N.; Frijling, J.L.; Veltman, D.J.; Mannens, M.M.A.M.; Olff, M. Oxytocin receptor gene methylation in male and female PTSD patients and trauma-exposed controls. Eur. Neuropsychopharmacol. 2019, 29, 147–155. [Google Scholar] [CrossRef]
- Ein-Dor, T.; Verbeke, W.J.M.I.; Mokry, M.; Vrtička, P. Epigenetic modification of the oxytocin and glucocorticoid receptor genes is linked to attachment avoidance in young adults. Attach. Hum. Dev. 2018, 20, 439–454. [Google Scholar] [CrossRef] [Green Version]
- Cappi, C.; Diniz, J.B.; Requena, G.L.; Lourenço, T.; Lisboa, B.C.G.; Batistuzzo, M.C.; Marques, A.H.; Hoexter, M.Q.; Pereira, C.A.; Miguel, E.C.; et al. Epigenetic evidence for involvement of the oxytocin receptor gene in obsessive-compulsive disorder. BMC Neurosci. 2016, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agorastos, A.; Pervanidou, P.; Chrousos, G.P.; Baker, D.G. Developmental Trajectories of Early Life Stress and Trauma: A Narrative Review on Neurobiological Aspects Beyond Stress System Dysregulation. Front. Psychiatry 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Hack, L.M.; Dick, A.L.W.; Provençal, N. Epigenetic mechanisms involved in the effects of stress exposure: Focus on 5-hydroxymethylcytosine. Environ. Epigenet. 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Yehuda, R.; Daskalakis, N.P.; Bierer, L.M.; Bader, H.N.; Klengel, T.; Holsboer, F.; Binder, E.B. Holocaust Exposure Induced Intergenerational Effects on FKBP5 Methylation. Biol. Psychiatry 2016, 80, 372–380. [Google Scholar] [CrossRef] [Green Version]
- Pang, T.Y.C.; Short, A.K.; Bredy, T.W.; Hannan, A.J. Transgenerational paternal transmission of acquired traits: Stress-induced modification of the sperm regulatory transcriptome and offspring phenotypes. Curr. Opin. Behav. Sci. 2017, 14, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Saavedra-Rodríguez, L.; Feig, L.A. Chronic social instability induces anxiety and defective social interactions across generations. Biol. Psychiatry 2013, 73, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Why I’m Sceptical about the Idea of Genetically Inherited Trauma. The Guardian, 11 September 2015. Available online: http://www.theguardian.com/science/blog/2015/sep/11/why-im-sceptical-about-the-idea-of-genetically-inherited-trauma-epigenetics (accessed on 16 May 2021).
- Birney, E. Chromatin and heritability: How epigenetic studies can complement genetic approaches. Trends Genet. TIG 2011, 27, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Relton, C.L.; Smith, G.D. Epigenetic Epidemiology of Common Complex Disease: Prospects for Prediction, Prevention, and Treatment. PLoS Med. 2010, 7, e1000356. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Gene | Reference | Species | Stressors | Epigenetic Changes |
|---|---|---|---|---|
| FKBP5 | Saito et al., 2020 [81] | Human | Childhood abuse | demethylation of FKBP5 intron 7 |
| Misiak et al., 2020 [82] | Human | Adverse childhood experiences (ACEs) | demethylation of FKBP5 | |
| Ramo-Fernandez et al., 2019 [83] | Human | Childhood maltreatment | demethylation of FKBP5 | |
| NR3C1 | Borcoi et al., 2021 [84] | Human | Food and nutritional security or insecurity status | hypermethylation of NR3C1 1F promoter |
| Pinheiro et al., 2021 [85] | Human | Alcohol consumption, Body mass index—BMI | hypomethylation of NR3C1 1F promoter (alcohol consumption), hypermethylation of NR3C1 1F promoter (BMI) | |
| Misiak et al., 2021 [86] | Human | Adverse childhood experiences | hypomethylation of NR3C1 | |
| BDNF | Duffy et al., 2020 [87] | Rat | Aversive caregiving | hypermethylation BDNF |
| Blaze et al., 2017 [88] | Rat | Caregiver maltreatment | hypermethylation BDNF IV | |
| Niknazar et al., 2017 [89] | Rat | Preconception maternal stress | hypermethylation BDNF | |
| SLC6A4 | Delano et al., 2021 [90] | Human | Maternal community-level deprivation | hypermethylation of 8 CpGs SLC6A4 |
| Anurag et al., 2019 [91] | Human | Early adversity (children of alcoholics) | hypermethylation of SLC6A4 | |
| Smith et al., 2017 [92] | Human | Adverse neighborhood environment | hypermethylation of SLC6A4 | |
| OXTR | Kogan et al., 2019 [93] | Human | Childhood adversity and socioeconomic instability | hypermethylation of OXTR |
| Kogan et al., 2018 [94] | Human | Childhood adversity | hypermethylation of OXTR | |
| Gouin et al., 2017 [95] | Human | Early life adversity (ELA) | hypermethylation of OXTR in females |
| Gene | Reference | Species | Stress-related disorder | Findings |
|---|---|---|---|---|
| FKBP5 | Mihaljevicab et al., 2021 [115] | Human | Psychotic disorder | (A) demethylation of FKBP5 (B) genetic background influence epigenetic modifications |
| Kang et al., 2018 [116] | Human | Post-Traumatic Stress Disorder (PTSD) | (A) demethylation of FKBP5 (B) genetic background influence epigenetic modifications | |
| Klinger-Konig et al., 2019 [117] | Human | Depression | (A) demethylation of FKBP5 associated with depression (B) genetic background influence epigenetic modifications | |
| Misiak et al., 2020 [82] * | Human | Psychotic disorders | (A) demethylation of FKBP5 associated with depression | |
| NR3C1 | Borcoi et al., 2021 [84] * | Human | Depressive symptoms | (A) hypermethylation of NR3C1 1F promoter |
| Pinheiro et al., 2021 [85] * | Human | Depression | (A) hypermethylation of NR3C1 1F receptor | |
| Bakusic et al., 2021 [118] | Human | Depression | (A) hypermethylation of NR3C1 | |
| Misiak et al., 2021 [86] * | Human | Various stages of psychosis | (A) hypomethylation of NR3C1 in first-episode psychosis patients (B) hypermethylation of NR3C1 in schizophrenia patients | |
| BDNF | Guo et al., 2021 [119] | Human | Post-Traumatic Stress Disorder (PTSD) | (A) hypermethylation of BDNF promoter (B) genetic background influence epigenetic modifications |
| Hossack et al., 2020 [120] | Human | Post-Traumatic Stress Disorder (PTSD) | (A) hypomethylation of BDNF promoter | |
| Peng et al., 2019 [121] | Human | Depression | (A) hypermethylation of BDNF promoter at 2 CpGs (B) hypomethylation of BDNF promoter at 1 CpG | |
| Shirata et al., 2020 [122] | Human | High sociotropy | (A) hypermethylation of BDNF promoter | |
| SLC6A4 | Sanwald et al., 2021 [123] | Human | Depression | (A) hypermethylation of SLC6A4 |
| Hossack et al., 2020 [120] | Human | Post-Traumatic Stress Disorder (PTSD) | (A) hypomethylation of SLC6A4 | |
| Peng et al., 2019 [121] | Human | Depression | (A) hypermethylation of SLC6A4 | |
| Schneider et al., 2017 [124] | Human | Major Depression Disorder (MDD) | (A) hypomethylation of SLC6A4 | |
| OXTR | Nawjin et al., 2018 [125] | Human | Post-Traumatic Stress Disorder (PTSD) | (A) hypermethylation of OXTR in females |
| Kogan et al., 2018 [94] | Human | Substance use problem | (A) hypermethylation of OXTR | |
| Ein-Dor et al., 2018 [126] | Human | Attachment avoidance | (A) hypermethylation of OXTR | |
| Cappi et al., 2016 [127] | Human | Obsessive-compulsive disorder | (A) hypermethylation of OXTR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mourtzi, N.; Sertedaki, A.; Charmandari, E. Glucocorticoid Signaling and Epigenetic Alterations in Stress-Related Disorders. Int. J. Mol. Sci. 2021, 22, 5964. https://doi.org/10.3390/ijms22115964
Mourtzi N, Sertedaki A, Charmandari E. Glucocorticoid Signaling and Epigenetic Alterations in Stress-Related Disorders. International Journal of Molecular Sciences. 2021; 22(11):5964. https://doi.org/10.3390/ijms22115964
Chicago/Turabian StyleMourtzi, Niki, Amalia Sertedaki, and Evangelia Charmandari. 2021. "Glucocorticoid Signaling and Epigenetic Alterations in Stress-Related Disorders" International Journal of Molecular Sciences 22, no. 11: 5964. https://doi.org/10.3390/ijms22115964
APA StyleMourtzi, N., Sertedaki, A., & Charmandari, E. (2021). Glucocorticoid Signaling and Epigenetic Alterations in Stress-Related Disorders. International Journal of Molecular Sciences, 22(11), 5964. https://doi.org/10.3390/ijms22115964